

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Glutamate is the major excitatory neurotransmitter in the central nervous system. Glutamatergic synaptic transmission has been implicated in learning and memory, and synaptic plasticity. However, high levels of extracellular glutamate play a hazardous role under certain pathological conditions, which is called glutamate toxicity [123]. In the mechanisms underlying glutamate toxicity, there are two pathways triggered by different types of membrane-bound receptors/transporters [4].
The first one, also called excitotoxicity, is triggered by the excessive activation of glutamate receptors including Ca2+-permeable N-methyl-D-aspartate receptors (NMDAR) and G protein-coupled metabotropic glutamate receptors (mGluR). The subsequent excessive Ca2+ influx from extracellular space was followed by mitochondrial overload of Ca2+ overload through mitochondrial Ca2+ channels such as mitochondrial calcium uniporter. The reciprocal interaction between mitochondrial Ca2+ (mtCa2+) and reactive oxygen species (mtROS) causes vicious mitochondrial dysfunction through a positive feedback mechanism [5].
The other pathway is mediated by cystine/glutamate antiporters in the cell membrane. Cystine uptake from the extracellular space is disturbed by high levels of extracellular glutamate, followed by glutathione (GSH) depletion. These processes lead to lipoxygenase activation and increase ROS levels [6789].
Both types of these pathways in glutamate toxicity mechanism result in the mitochondrial overload of Ca2+ and ROS via several cellular mechanisms, which is crucial in determining the fate of neuronal cell survival and death [235]. Mitochondrial overload of Ca2+ and ROS results in mitochondrial membrane potential (ΔΨm) disruption, a second boost of ROS generation in mitochondrial electron transport chains (ETC), adenine triphosphate (ATP) depletion, and mitochondrial membrane permeabilization (MMP). Pro-apoptotic Bcl-2 family members-mediated Bak/Bax oligomerization evokes MMP in the outer membrane [7891011]. Proapoptotic factors such as cytochrome c and apoptosis-inducing factor are released from the mitochondrial intermembrane space through MMP pores and finally cause mitochondria-dependent cell death [12131415]. Neuronal cell death inevitably leads to dysfunction of the nervous system and therefore has been implicated in brain ischemia/stroke, traumatic brain injury, and neurodegenerative diseases [161718].
Imbalance between intracellular ROS and cellular anti-oxidant enzyme system results in mitochondrial dysfunction and neuronal cellular death against glutamate toxicity. Heme oxygenase-1 (HO-1) plays an important anti-oxidant enzyme which detoxifies ROS through the formation of biliverdin and bilirubin, both of which are exerted as ROS scavengers [19]. Nuclear factor-like 2 (Nrf2), a basic leucine zipper transcription factor, is basally sequestered by kelch-like protein 1 (Keap1). However, in response to certain stimuli, the phosphorylated Nrf2 is released from Keap1 and then translocates to the nucleus where it activates the anti-oxidant response element in the promoter region of several anti-oxidant genes such as HO-1 [2021]. Therefore, Nrf2/HO-1 axis as a cellular anti-oxidant defense system has been extensively investigated in ROS-related neuronal insult models.
Several research groups have shown that dieckol (DEK), one of the phlorotannins isolated from the marine brown alga Ecklonia cava , exhibits multifunctional biological activities such as anti-oxidant, anti-inflammatory, and anti-cancer activities in diverse cell types [2223] as well as improved cognitive function in ethanol-treated mice [24]. We recently reported that DEK inhibits lipopolysaccharide-stimulated microglia activation and subsequent microglia-mediated neuronal cell death via mitogenactivated protein kinases, Akt, and nicotinamide adenine dinuclelotide phosphate (NADPH) oxidase-mediated pathways [25]. We demonstrate here that DEK exerts neuroprotective activities in glutamate-stimulated neurons against mitochondria-dependent neuronal cell death through its direct free radical scavenging property and the Nrf2/HO-1 pathway activation.
METHODS
Reagents
5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethylbenzimidazolocarbocy anine iodide (JC-1), tetramethylrhodamine, ethyl ester, rhod-2 acetoxymethyl ester (Rhod-2 AM), MitoT racker Green, MitoSOX Red, ATP determination kit, and reagents used in cell culture were obtained from Invitrogen (Carlsbad, CA, USA). Antibodies against Nrf2, HO-1, and β-actin were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). The a ntibody against the TATA binding protein (TBP) was obtained from Abcam Plc. (Abcam, Cambridge, UK). Horseradish peroxide-conjugated immunoglobulin G antibody was purchased from Vector Laboratories (Burli ngame, MA, USA). The lactate dehydrogenase release (LDH) cytotoxicity detection kit was obtained from Takara Shuzo Co. (Shiga, Japan). All other reagents were purchased from Sigma (St. Louis, MO, USA), unless indicated.
Extraction and isolation of DEK
As previously described [25], the whole plant of the marine brown alga Ecklonia cava was prepared from the Jeju Island coast in the Republic of Korea. Briefly, the dried power of Ecklonia cava was extracted with 70% aqueous ethanol. The n-butanol fraction was performed by octadecyl silica gel column chromatography. The DEK was finally purified by LH-20 column chromatography and then confirmed by comparing the mass spectrometry, 1H-nuclear magnetic resonance (NMR), and 13C-NMR data [26].
Cell culture
Primary cortical neuron cultures were prepared from the cerebral cortices of gestation day 16 Sprague-Dawley rat embryos. The meninges-free rat cortices were triturated using fire-polished Pasteur pipettes. Cells were seeded into culture plates pre-coated with poly-L-lysine and cultured in Minimum Essential Media (Gibco BRL, Gaithersburg, MD, USA) containing 10% fetal bovine serum (FBS), 45% glucose, 25 mM glutamate, 100 mM sodium pyruvate, 100 U/ml penicillin, and 100 µg/ml streptomycin and kept at 37℃ in a humidified atmosphere of 5% CO2. After 24 h incubation, the culture medium was replaced by neurobasal media supplemented with 2% B27 supplement, 200 mM L-glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin. The HT22 neuronal cell line [27] was generously supplied by Dr. B.H. Lee (Gachon University of Medicine and Science, Republic of Korea). HT22 cells were cultured in Dulbecco's modification of Eagle medium (Gibco BRL, Gaithersburg, MD, USA) supplemented with 10% FBS, 100 U/ml penicillin, and 100 µg/ml streptomycin and maintained at 37℃ in a humidified atmosphere of 5% CO2. The study was approved by the Animal Care and Use Committee of Jeju National University (no. 2016-0029) and all experiments were performed in accordance with the guidelines.
1,1-Dip henyl-2-picrylhydrazyl (DPPH) free radical scavenging assay
As previously described [25], 10 µl of DEK in various concentrations was added to 190 µl of 0.15 mM DPPH, and mixed vigorously. The mixture was incubated in the dark at room temperature for 2 h, and the absorbance was read at 517 nm using a microplate reader (Sunris e; Tecan, Mannedorf, Switzerland).
Intrace llular ROS measurement
As previously described [25], primary cortical neurons and HT22 neurons were seeded in 96-well tissue culture plates at 2 × 104 cells/well containing medium (200 ml) after 12 h stabilization. After the addition of 20 µM of 2′,7′-dichlorofluorescein diacetate (DCF-DA), the fluorometric analysis was performed with the excitation/emission wavelength at 485 nm/535 nm using a microplate reader (Spectra fluor; Tecan).
LDH assay
The cytotoxicity of DEK was measured with an LDH cytotoxicity detection kit (Takara Shuzo Co.) on a mi croplate reader (Sunrise), as previously described [28].
MTT cell viability assay
MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) was used to investigate the DEK effects on cell viabilities as previously described [28].
Quantif ication of cellular ATP levels
After treatment, the cells were harvested and then lysed with lysis buffer (120 mM NaCl, 40 mM Tris [pH 8], 0.1% NP 40) and centrifuged at 13,000 × g for 15 min. The supernatants were assayed for ATP content using luciferase/luciferin ATP determination kit (Molecular Probes, Eugene, OR, USA).
Western blot analysis
Western blot analysis was performed as described [28]. The protein bands were detected using an enhanced chemiluminescence Western blot detection kit (Amersham, Little Chalfont, UK). Nuclear proteins were prepared as previously described [28]. The membranes were probed with the following primary antibodies: HO-1 (1:1,000), Nrf2 (1:200), TBP (1:2,000), and β-actin (1:5,000). β-actin and TBP were used as each control for equal protein loading in whole cells and nuclear fractions, respectively.
Flow cy tometric analysis of ΔΨm, mitochondrial Ca2+ and ROS
JC-1 is a cell-permeable indicator for ΔΨm and membrane potential-dependently accumulated in active mitochondria. Rhod-2 AM, a cell-permeable AM form of Rhod-2, was used as a mitochondria-selective Ca2+ indicator. Upon Ca2+ binding, fluorescence intensities of Rhod-2 are dramatically increased (> 100-fold). As cationic Rhod-2 shows potential-driven uptake into the mitochondria, it has been used as a selective indicator for mitochondrial Ca2+. The cationic MitoSOX Red is cell-permeable and also selectively targeted to the mitochondria. Once MitoSOX Red is oxidized by superoxide anions which are the predominant ROS in mitochondria, it exhibits red fluorescence. After treatment, the cells were loaded with JC-1 (5 µM, 30 min), Rhod-2 AM (2 M, 30 min) and MitoSOX Red (5 µM, 20 min) at 37℃ to measure ΔΨm, mitochondrial Ca2+ and ROS, respectively [2930]. The loaded cells were washed with phosphate-buffered saline three times, and analyzed by flow cytometry using a FACS Calibur (Becton Dickinson, Franklin Lakes, NJ, USA).
Confocal imaging analysis of mitocho ndrial ROS
After trea tment, HT22 neuro ns were loa ded with the mitochondrial superoxide indicator MitoSOX Red (5 µM, 20 min) and Mito Tracker Green (50 nM, 30 min), washed thr ee times and imaged on a confocal laser scanning microscope (FV500; Olympus, Tokyo, Japan) using a cooled charge-coupled device camera controlled by Flow View 4.2 software (Olympus).
RESULTS
Neuroprotective effects of DEK on neuronal cell viability and morphological changes against glutamate toxicity
We investigated whether DEK affected neuronal cell viability against glutamate toxicity. Both primary cortical neurons and HT22 neuronal cell line were pretreated with DEK for 1 h prior to glutamate stimulation. Then, primary cortical neurons were stimulated with glutamate (100 µM) in the presence of different doses (1, 10, 30 and 50 µM) of DEK for 24 h. In case of HT22 neurons, cells were stimulated with glutamate (5 mM) for 12 h. DEK below 100 µM did not show any cytotoxicity as a chemical agent for treatment, based on MTT assay and LDH release assay (Fig. 1B, C).
The results revealed that DEK markedly increased neuronal cell viability against glutamate toxicity in a dose-dependent manner in both the primary cortical neurons (Fig. 2A) and HT22 neurons (Fig. 2B). Glutamate-induced morphological changes were observed by phase contrast inverted microscopy. Retracted neurites and shrunken cell bodies were shown in the glutamate-stimulated HT22 neurons, and DEK recovered glutamate-induced morphological deterioration (Fig. 2C). Taken together, these results suggest that DEK significantly protects glutamate-stimulated neurons against glutamate toxicity-induced cell death and deterioration.
ROS scavenging activities of DEK in glutamate-stimulated neurons
We investigated using the DPPH free radical scavenging assay whether DEK had direct free-radical scavenging properties in a cell-free system. As shown in Fig. 3A, the DEK dose-dependently scavenged DPPH free radicals. Both primary cortical neurons and HT22 neuronal cell line were pretreated with DEK for 1 h prior to glutamate stimulation. Then, primary cortical neurons were stimulated with glutamate (100 µM) in the presence of different doses (1, 10, 30, 40, and 50 µM) of DEK for 24 h. In case of HT22 neurons, cells were stimulated with glutamate (5 mM) for 12 h. The amounts of intracellular ROS generated by glutamate were detected by a spectrofluorometer using the ROS-sensitive fluorescent dye DCF-DA. The results demonstrated that glutamate markedly evoked intracellular ROS generation in both the primary cortical neurons (Fig. 3B) and HT22 neurons (Fig. 3C), and DEK strongly suppressed glutamate-induced intracellular ROS levels in a dose-dependent manner in both types of neurons.
DEK attenuates glutamate-induced mitochondrial dysfunction
Mitochondrial dysfunction leads to mitochondria-dependent cell death against glutamate toxicity, which includes mitochondrial Ca2+ overload, ΔΨm disruption, ATP depletion, abnormal electron transport, and ROS generation in the mitochondrial ETC. Therefore, we investigated whether DEK protected neurons from mitochondrial dysfunction in several mitochondrial parameters. Intracellular ATP levels were measured using a luciferase/luciferin ATP determination kit. ΔΨm, mitochondrial Ca2+, and ROS were measured using by flow cytometric analysis of JC-1, Rhod-2, and MitoSOX fluorescence, respectively.
HT22 neurons were pretreated with DEK (50 µM) for 1 h, then stimulated with glutamate (5 mM) in the presence of DEK for 12 h. As shown in Fig. 4, DEK significantly rescued glutamate-induced ATP depletion (Fig. 4A), ΔΨm disruption (Fig. 4B), mitochondrial Ca2+ overload (Fig. 4C), and mitochondrial ROS generation (Fig. 4D). The inhibitory effects of DEK on mitochondrial ROS were also shown in confocal microscopic images (Fig. 4E). Dramatic accumulation of mitochondrial ROS stained with MitoSOX was visualized in a dotted pattern under confocal microscopy. Taken together, these results suggest that DEK significantly protects HT22 neurons against glutamate-induced mitochondrial dysfunction.
DEK increases nuclear translocation of Nrf2 and HO-1 expression
HO-1 plays an important anti-oxidant enzyme which detoxifies ROS and the phosphorylated Nrf2 translocates to the nucleus where it activates HO-1 gene transcription [192021]. Therefore, the effects of DEK on the Nrf2/HO-1 axis as a cellular anti-oxidant defense system were investigated in HT22 neurons.
The cells treated with DEK (10, 20, 30, 40, and 50 µM) for 12 h showed a dose-dependent increase in HO-1 protein expression (Fig. 5A). In addition, DEK dramatically increased protein expression levels of the transcription factor Nrf2 in the nuclear fraction, suggesting DEK-induced nuclear translocation of Nrf2. The maximal increase was shown at the 90 min time-point (Fig. 5B). Taken together, these results suggest that DEK markedly reduced intracellular ROS levels (Fig. 3B, C) against glutamate toxicity through the activation of the Nrf2/HO-1 pathway (Fig. 5) as well as direct free radical scavenging activity (Fig. 3A).
DISCUSSION
The neuroprotective mechanisms of DEK against glutamate-induced mitochondrial dysfunction and neuronal cell viability against glutamate toxicity were investigated in this study. The results demonstrated that DEK exerts neuroprotective activities through direct free radical scavenging and Nrf2/HO-1 pathway activation against glutamate-induced mitochondrial dysfunction and subsequent mitochondria-dependent neuronal cell death.
Glutamate toxicity has been implicated in the pathogenesis of brain ischemia/stroke, traumatic brain injury, and neurodegenerative diseases such as Parkinson's, Alzheimer's, and Huntington's disease [161718]. Therefore, the glutamate toxicity model has long been employed in these research fields. Two different types of neurons were used to induce a glutamate toxicity model in this study. Rat primary cortical neurons were stimulated with glutamate (100 µM,) in the presence of DEK for 24 h. In case of mouse hippocampal HT22 neuronal cell line, cells were stimulated with glutamate (5 mM) for 12 h. The dose and duration of glutamate treatment were determined depending upon cell types, based on the previous reports [313233]. There are several different methods used to induce neuronal glutamate toxicity. The experimental conditions may differ according to the nature and strength of neurotoxic stimuli, neuronal cell types or particular populations. The differences/discrepancies in experimental conditions such as dose and duration of glutamate treatment between two types of cells, primary cortical neurons and HT22 neuronal cell line, are attributed to different expression pattern of glutamate receptors, especially NMDAR. HT22 cells lack in functional ionotropic glutamate receptor including NMDAR, compared to primary cultured neurons, and therefore need more glutamate stimulation (i.e., 5 mM) to induce glutamate toxicity [3233].
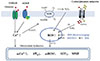
The two different pathways underlying glutamate toxicity mechanism are integrated in parallel in primary cultured neurons; NMDAR and/or mGluR-mediated excitotoxicity pathway and nonreceptor (i.e., cystine/glutamate antiporters)-mediated oxidative stress pathway (Fig. 6). HT22 cell system has been used as an excellent model for nonreceptor-mediated oxidative stress by glutamate. Independently of NMDAR activation, cystine uptake from extracellular space is disturbed by high levels of extracellular glutamate in the brain, followed by GSH depletion [2733]. In this study, we demonstrated that DEK exerts neuroprotective effects against glutamate toxicity in these two different cell systems. However, this study was not focused on either comparison or differences between two different cell models. Most experiments such as mitochondrial dysfunction and Nrf-2/HO-1 expression were accomplished in HT22 neuronal cell line. Only neuroprotective effects of DEK on cell viability and intracellular ROS levels against glutamate toxicity were also confirmed in primary cortical neurons, as well as in HT22 cells.
ROS serve as signaling molecules in certain redox-sensitive pathways in response to several extracellular stimuli under physiological conditions. For example, H2O2, one of the ROS best characterized as a signaling molecule, inactivates protein tyrosine phosphatases by oxidizing cysteine residues and thereby regulates protein tyrosine phosphorylation [19]. Localized ROS accumulation is tightly regulated according to cellular needs. However, high levels of ROS, if global and sustained, evoke mitochondria-dependent neuronal cell death. Therefore, cellular ROS levels are precisely controlled by cellular antioxidant defense mechanisms including catalase, superoxide dismutase [34], and HO-1 [19], and a complex intracellular network of enzymes such as GSH, GSH peroxidase, peroxiredoxin, thioredoxin, and thioredoxin reductase [35]. Nevertheless, imbalance between ROS and antioxidants inside the cells results in mitochondrial dysfunction and neuronal cellular death.
Intracellular ROS levels are markedly increased in glutamate-stimulated neurons through both glutamate receptor-mediated excitotoxicity pathway and nonreceptor-mediated oxidative glutamate toxicity pathway. It has been established that NADPH oxidase is an important down-stream protein of the glutamate receptors. The Ca2+ influx through NMDAR is linked to the activation of phosphoinositide 3 kinase (PI3K), followed by atypical protein kinase C zeta (PKC zeta) [3637]. Extensive NMDAR activation leads to NADPH oxidase-induced superoxide formation by the phosphorylation of the cytosol subunit p47phox mediated by PI3K and atypical PKC zeta. Therefore, NMDAR and/or mGluR-mediated intracellular Ca2+ influx and NADPH oxidase-dependent ROS are evaluated as triggering events in the mechanisms underlying glutamate receptor-mediated excitotoxicity [43637].
Imbalance between ROS and cellular anti-oxidant enzyme system results in mitochondrial dysfunction and neuronal cellular death against glutamate toxicity. We demonstrated that DEK markedly reduced intracellular ROS levels (Fig. 3B, C) and mitochondrial ROS levels (Fig. 4D, E), against glutamate toxicity. DEK showed direct free radical scavenging activity in a cell-free system, as shown in DPPH assay (Fig. 3A). In addition, we revealed that DEK activated the Nrf2/HO-1 axis as a cellular anti-oxidant defense system in HT22 neurons (Fig. 5). Taken together, these results suggest that neuroprotective mechanisms of DEK against glutamate toxicity are largely based on the reduction of ROS levels through the activation of the Nrf2/HO-1 pathway (Fig. 5) as well as direct free radical scavenging activity (Fig. 3A).
As shown in Fig. 6, glutamate toxicity-induced increase of intracellular ROS levels finally induces mitochondrial dysfunction, which includes mitochondrial overload of Ca2+, and ROS, ΔΨm disruption, a second boost of ROS generation in ETC, ATP depletion. It is suggested that the action sites of DEK may be intracellular, as shown in Fig. 6. DEK has multiple phenol ring structures. Based on the chemical structure (Fig. 1A), it is possible that DEK possess both hydrophobic and hydrophilic properties. Multiple benzenoid ring structures underlie the hydrophobic property of DEK while hydroxy (−OH) functional groups may allow DEK hydrophilic. Regarding this issue, it was reported that rhodamine-labelled DEK was observed inside the cells on a fluorescence microscope, when treated outside the cells [38]. Taken together, it is suggested that the hydrophobic DEK may rapidly diffuse across cell membranes so that they can reach intracellular sites of action.
In summary, we demonstrated here that DEK exerted neuroprotective activities against glutamate toxicity in both the primary cortical neurons and HT22 neurons through direct ROS scavenging and activation of the Nrf-2/HO-1 pathway as a cellular anti-oxidant defense system. The results suggest that DEK may be a promising neuroprotective agent against glutamate toxicity-related conditions implicated in brain ischemia/stroke, traumatic brain injury, and neurodegenerative diseases.





 XML Download
XML Download