

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Cardiac amyloidosis is defined as an abnormal cardiac deposit of proteins that are not native to the heart, generally insoluble, and resistant to proteolysis.1) These proteins accumulate in the extracellular space of the myocardium, more accurately in the subsarcolemma, and lead to direct cell toxicity, cell death, and expansion of the extracellular space. Clinically, deposition of amyloid proteins in the heart causes thickening of the ventricular wall, diastolic dysfunction, and eventually, heart failure and sudden cardiac death. Over 10 proteins have been identified to cause cardiac amyloidosis to date; however, the majority of abnormal proteins frequently encountered are light chain immunoglobulin (AL), wild type transthyretin (wtTTR), and mutant transthyretin (mtTTR). An amyloid protein is noted with a letter ‘A’ in front of the name. For example, the amyloid form of transthyretin (TTR) is noted as ATTR and the amyloid light chain as AL.
Cardiac amyloidosis is a rare disease. In the US, the prevalence of AL type is estimated to be approximately 1 in 10 million2)3) and is almost always associated with plasma cell dyscrasia. Although the prevalence of ATTR cardiac amyloidosis has never been investigated systematically, there have been reports demonstrating that wtTTR cardiac amyloidosis might be underestimated in the elderly population4); its prevalence has been reported to be up to 25% in those 80 years or older.5) In line with others,6)7) we also found that there is a group of patients with incidental cardiac amyloidosis and severe aortic stenosis, and that this is linked to ventricular dysfunction, suggesting possible clinical relevance of ‘incidental’ cardiac amyloidosis.
DIAGNOSIS OF CARDIAC AMYLOIDOSIS
The gold standard diagnostic method of cardiac amyloidosis is identification of abnormal proteins deposited abundantly in the heart to cause organ dysfunction. Before invasive tools are employed for definite diagnosis, there are certain clues that indicate clinical suspicion of cardiac amyloidosis. Cardiac amyloidosis is often diagnosed late, with an average time from initial symptoms to definite diagnosis of at least 6 months.8) This results in significant delays in appropriate treatment, which highlights the importance of clinical suspicion. We will list some typical findings in noninvasive tests with relevant references. The clinical presentation of cardiac amyloidosis may be very diverse; therefore, absence of the listed findings does not preclude disease absence.
12-LEAD ELECTROCARDIOGRAM
An abnormal finding occurs in up to 90% of patients with cardiac amyloidosis.9) A typical 12-lead electrocardiogram finding in patients with cardiac amyloidosis is a low QRS voltage in the limb leads, especially in spite of increased left ventricular (LV) wall thickness (Figure 1A).10) Low voltage QRS is extremely rare in other diseases that frequently present with a thickened LV wall such as hypertrophic cardiomyopathy, aortic stenosis, or hypertension (Figure 1B). The presence of low QRS voltage portends a worse outcome in cardiac amyloidosis patients.11) A pseudoinfarction pattern, defined as QS waves in any two consecutive leads, is also a common finding (Figure 1C). Interestingly, these findings may be present irrespective of increased ventricular wall thickness. These findings may be even more prevalent in patients without increased ventricular wall thickness.10)
Figure 1
Examples of 12-lead electrocardiograms of cardiac amyloidosis patients. (A, B) Two patients with a similar degree of LV wall thickness, 15 mm. A is a case of cardiac amyloidosis, and B is a case of the concentric form of hypertrophic cardiomyopathy. (A) Despite increased ventricular wall thickness, there is no evidence of ventricular hypertrophy on electrocardiograms. The QRS voltage, especially in the limb leads and the lateral precordial leads, is rather low considering the presence of thickened left ventricular wall. (B) This is in contrast to a significant increase of QRS voltage in patients with hypertrophic cardiomyopathy. (C) A pseudoinfarction pattern in a patient with cardiac amyloidosis. A typical QS wave (red arrows) can be seen in the septal precordial leads and can be misdiagnosed as myocardial infarction.

The above listed electrocardiographic findings are found more commonly in those with the AL type than the ATTR type. Despite these findings, the prevalence of typical electrocardiographic findings does not exceed 50%; thus, absence of low QRS voltage or the pseudoinfarction pattern does not exclude the possibility of cardiac amyloidosis.
ECHOCARDIOGRAPHY
Cardiac amyloidosis should be suspected in any patient with symptoms or signs of unexplained heart failure, especially when LV wall thickness is increased and when there is prominent diastolic dysfunction (Figures 2A and B). Typically, cardiac amyloidosis may present as concentric thickening of the ventricular wall as opposed to hypertrophic cardiomyopathy, in which localized ventricular thickening is more common. Before harmonic imaging was commonly used, the ventricular wall of cardiac amyloidosis patients was described as having a ‘sparkling or speckled appearance,’ but this finding is now considered obsolete.12) Also, cardiac amyloidosis may be present without thickening of the LV wall.13) Additionally, valvular thickening or pericardial effusion may also be evident with biatrial enlargement that reflects chronic diastolic dysfunction (Figure 2C). Diastolic dysfunction, increased wall thickness, atrial enlargement, and pericardial effusion are all independent prognosticators in patients with cardiac amyloidosis.9)14)15)16) Although the degree of ventricular wall thickening may be more severe in those with ATTR than in those with AL cardiac amyloidosis,17) direct comparison of ventricular structure and function between ATTR and AL cardiac amyloidosis remains to be confirmed in a larger cohort.
Figure 2
Typical echocardiography features in cardiac amyloidosis patients. (A) Thickened LV wall that measures up to 14 mm on parasternal view. (B) Doppler interrogation demonstrates a restrictive filling pattern in the mitral inflow study and very low e′ velocity at the mitral septal annulus. Calculation of E/e′ is estimated to be 30, which indicates high LV filling pressure. (C) An angina patient who developed cardiac amyloidosis after 5 years of percutaneous coronary intervention. The LV wall thickness at the time of coronary intervention was normal, as were the sizes of both atria (left panel). The patient visited the hospital for progressive dyspnea and significant thickening of the ventricular wall, and biatrial enlargement was noted (right panel). Note thickening of the tricuspid valve at the time of diagnosis of cardiac amyloidosis (yellow arrows) that was not evident on initial echocardiography. (D) A representative bull's eye plot of longitudinal strain. Note that the longitudinal strain of the apex is preserved in contrast to those of the other midventricular or basal segments.

Perhaps the most up-to-date echocardiographic modality used for diagnosis and risk stratification of cardiac amyloidosis is strain imaging, generally using speckle tracking echocardiography. A typical strain image of a patient with cardiac amyloidosis is an ‘apical sparing’ or a ‘cherry-on-top’ pattern on the bull's eye plot of global longitudinal strain (Figure 2D).18) Although not validated in other studies, this apical sparing pattern was shown to have 93% sensitivity and 82% specificity for diagnosing cardiac amyloidosis, and its visual assessment is useful for diagnosing less advanced stages of cardiac amyloidosis.19) Longitudinal ventricular dysfunction by strain imaging, whether at the basal level or globally, signals poor prognosis in those with AL cardiac amyloidosis.16)20)21) The prognostic value of strain imaging in ATTR cardiac amyloidosis remains to be investigated in further studies.22)
CARDIOVASCULAR MAGNETIC RESONANCE
Cardiovascular magnetic resonance (CMR) is a versatile imaging method for analyzing myocardial texture and has been rapidly adopted for clinical use in patients with suspected cardiac amyloidosis. This is based on the seminal discovery of the difference in gadolinium kinetics of the myocardium between normal and amyloidosis patients.23)
The pattern of late gadolinium enhancement (LGE) in the myocardium is heterogeneous, ranging from the global transmural or subendocardial LGE pattern to patchy focal LGE to suboptimal myocardial nulling (Figure 3A), which may match the deposition pattern of interstitial amyloid.24) Incidental findings such as atrial enhancement (Figure 3B) or thrombus (Figure 3C) may also be detected on CMR, given its superiority in signal-to-noise ratio to echocardiography.
Figure 3
Cardiovascular magnetic resonance (CMR) images of cardiac amyloidosis patients. (A) The pattern of late gadolinium enhancement (LGE) can be diverse in cardiac amyloidosis patients. Although multifocal patchy LGE (A-1) and global subendocardial ring enhancement patterns (A-2) are most common, suboptimal nulling is not unusual (A-3) and even no LGE (A-4) in spite of cardiac amyloidosis on myocardial biopsy. Pericardial effusion is also noted (A-3, yellow arrows). (B) Along with subendocardial LGE in the ventricle, LGE may also be seen in the interatrial septum. (C) Analysis for the presence of intracardiac thrombi in patients with cardiac amyloidosis is important, as in this case in which thrombi were noted at both atria (yellow arrows). (D) Parametric CMR is easy to quantify and gives information on diffuse myocardial change. A typical case of cardiac amyloidosis with an elevated septal native T1 up to 1,550 msec (left panel). A CMR scan of a normal volunteer with a native T1 of 1,150 msec (right panel).

Based on the histologic background that amyloid is deposited in the interstitium, several investigators have looked into the possibility of using parametric CMR for assessment of cardiac amyloidosis, the utility of which has been demonstrated elsewhere.25)26) Either by measurement of native T1 or by calculation of extracellular volume (ECV) fraction (Figure 3D), the myocardium of cardiac amyloidosis patients has been shown to have higher native T1 or ECV.27)28) Although the majority of these studies have used specialized sequences for T1 mapping, such as modified look-locker inversion recovery (MOLLI) sequence or shortened MOLLI (shMOLLI) sequence, this strategy can also be adopted with the post-contrast conventional SSFP sequence that is used for cine imaging.29) Recent investigations have shown that the presence of LGE on CMR is a robust predictor of outcome,30) independent of other known cardiac biomarkers.31) The degree of myocardial edema, measured with the use of T2 imaging, has also been shown to be an independent prognosticator.32)
The validity of CMR in ATTR cardiac amyloidosis is not as well established as in AL cardiac amyloidosis, but there is no evident reason that the clinical validity of CMR in the ATTR type would be different from that in the AL type. However, the number of patients studied is much smaller, and most of the investigations are single-center studies. Nonetheless, the pattern of ventricular hypertrophy that can be precisely evaluated using cine CMR and the degree of ECV may be useful for evaluation of ATTR cardiac amyloidosis.33) Although unvalidated in larger populations, the pattern of LGE also may be used for differentiation between AL and ATTR34)35) amyloidosis.
In spite of the accumulating evidence of the value of CMR in diagnosis, differentiation, and prognostication of cardiac amyloidosis, there are some limitations. Renal dysfunction, which is commonly seen in systemic amyloidosis patients, is a contraindication for gadolinium use. Furthermore, diagnosis of cardiac amyloidosis with CMR relies on the pattern of LGE and, as such, the false-positive rate of up to 5% cannot be overlooked.34)
NUCLEAR SCINTIGRAPHY
Since the initial finding that 99mTc-diphosphonates can image amyloid deposits in the soft tissue,36) bone imaging tracers that are based on diphosphonates tagged with 99mTc, such as 99mTc-pyrophosphate (99mTc-PYP), 99mTc-methylene diphosphonate (99mTc-MDP), 99mTc-hydroxymethylene diphosphonate (99mTc-HPD), and 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid (99mTc-DPD), have long been used for visualization of amyloid deposits in the heart (Figure 4A). However, the theoretical basis of why these single-photon emission computed tomography (SPECT) agents bind to TTR is still not well understood. Of these agents, 99mTc-DPD has been the most extensively studied. The binding of 99mTc-DPD to amyloid deposits in the heart seems to be more specific to the ATTR type than the AL type.37)38) The binding of 99mTc-DPD does not seem to differentiate between wtTTR and mtTTR, as shown in a report of a series of mtTTR-related cardiac amyloidoses.39) Moreover, quantification of tracer retention in the myocardium reflects the severity of myocardial involvement.40)
Figure 4
Nuclear imaging in diagnosis of cardiac amyloidosis and follow-up. (A) A representative image of 99mTc-DPD SPECT in an ATTR cardiac amyloidosis patient. Evident uptake of 99mTc-DPD in the myocardium is noted and is supported by suboptimal nulling on CMR. (B) Uptake of 11C-Pittsburgh B (PiB) compound in a patient with biopsy-proven cardiac amyloidosis (left panel). This compound is specific for amyloid deposits as no 11C-PiB is noted in healthy volunteers (right panel). (C) A significant decrease of 11C-PiB uptake in a patient with cardiac amyloidosis 12 months after chemotherapy and autologous stem cell transplantation (left panel; before chemotherapy, right panel).
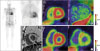
Contrary to 99mTc-diphosphonate SPECT tracers, recently developed positron emission tomography (PET) tracers are made to directly target the amyloid protein itself and stem from the findings in patients with Alzheimer's disease, a disease of amyloid accumulation in the brain parenchyme.41)42) We and others were among the first to report the clinical utility of 11C-Pittsburgh B (PiB) compound in imaging cardiac amyloidosis, albeit with some differences in acquisition protocol (Figure 4B).19)43) The 11C-PiB compound also has the potential to monitor the degree of amyloid deposition during or after appropriate chemotherapy (Figure 4C), but evidence supporting this is largely lacking. Although the experience in our center is limited, a few investigators have also reported use of 11C-PiB in ATTR cardiac amyloidosis.44)
However, because of the short half-life of 11C (t1/2≈20minutes) and the need for an on-site cyclotron, the majority of amyloid PET tracers is now labeled with18F-tagged radiotracers such as 18F (t1/2≈110minutes), 18F-florbetapir, or 18F-florbetaben. Following initial successful pilot studies using these 18F PET tracers,45)46) these 18F-tagged radiotracers have been shown to accumulate specifically at sites of amyloid deposit.47) Although not specific for amyloid, recent reports have shown that 18F-NaF also may be used for imaging cardiac amyloidosis.48)
CONCLUSION
Although cardiac amyloidosis is not a common disease, the diverse presentation often leads to misdiagnosis and inappropriate delays in adequate treatment. Therefore, clues that point toward diagnosis of this disease are important. Various imaging studies are utilized to clarify suspicion, to diagnose the disease noninvasively, and possibly for follow-up of the disease course.
Not all of the aforementioned studies may be needed in every patient. There are some pitfalls to consider in each test, some of which have been covered in this review. However, we believe that careful consideration of these tests is needed in patients with suspected or diagnosed cardiac amyloidosis, so that each patient will receive the optimal, tailored care needed.
 XML Download
XML Download