

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Background
Cases of drug-induced torsade de Pointes (TdP) that were reported in the 1990s and 2000s resulted in the market withdrawal of many drugs.[1] Because QTc interval prolongation by blocking the cardiac hERG channel was reported to provoke TdP,[2] in vitro studies to estimate hERG channel blockage and to provide thorough clinical trials measuring QTc (TQT studies) according to the ICH S7B and E14 guidelines have been a required part of drug development for the past decade.[3]
However, the issue of false-positive TQT study results was brought up a few years later.[4] Moreover, the correlation between the degree of hERG channel blockage at the Cmax relative to the IC50 (Cmax/IC50 ratio) and TdP risk was reported to include many outlier drugs, so that the hERG channel alone seemed inappropriate for use as a reliable screening criterion.[5]
In this context, approaches to improve the current practices of in vitro and clinical studies were added to the Q&A part of ICH guideline E14 (e.g., concentration–response modeling of TQT study data). The comprehensive in vitro proarrhythmia assay (CiPA) initiative was launched following a workshop at the US FDA in 2013 to improve assessment of the proarrhythmic potential of new drugs. Currently, members of the CiPA initiative include the regulators FDA, European Medicines Agency, Health Canada, Japan's Pharmaceuticals and Medical Devices Agency, industry, and academia. The initiative also coordinates with several organizations such as the Health and Environmental Science Institute, the Safety Pharmacology Society, and the Cardiac Safety Research Consortium. CiPA has four working groups dealing with ion channels, in silico models, myocytes, and regulatory guidelines (Fig. 1), and each of these groups has been working to accomplish various milestones that are to be finalized by the end of 2018. In this report, these four working group components will be introduced briefly, along with current results and their implications.
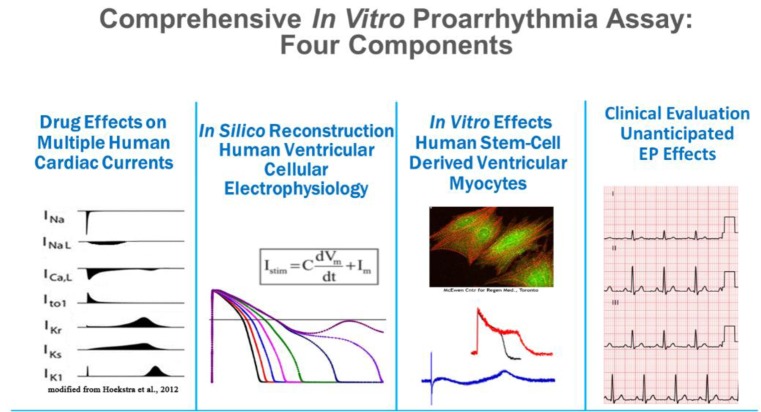 | Figure 1Four components of CiPA: the ion channel, in silico, cardiomyocyte, and ECG biomarker working groups (http://cipaproject.org/about-cipa/#WorkStreams).
|
Go to : 
Limitations of the current hERG blockage and QTc prolongation paradigm
This section introduces two typical drugs (verapamil and ranolazine) that block hERG channels and prolong the QTc, but do not cause TdP. New molecules of this kind would be screened for simple hERG blockage and weeded out according to the current guidelines. Even if they enter clinical development, prolonged QTc findings in TQT studies will raise serious concerns for their further development.
Both verapamil and ranolazine bind to the hERG ion channel and cause prolonged action potential duration (APD) and, thus, QTc. Although the APD is prolonged, the shape of the AP may be somewhat different from those produced by QTc prolonging drugs that cause TdP (Fig. 2). As blocking inward currents (calcium by verapamil, late sodium by ranolazine) prevents the occurrence of early afterdepolarizations (EADs), the risk of TdP is not increased by verapamil or ranolazine. If these two drugs had been tested for hERG channel blockage and QTc prolongation, they must have been discarded before further development. In the CiPA project, efforts were made to discover biomarkers that could help to discern the false-positive (+) TdP risks.
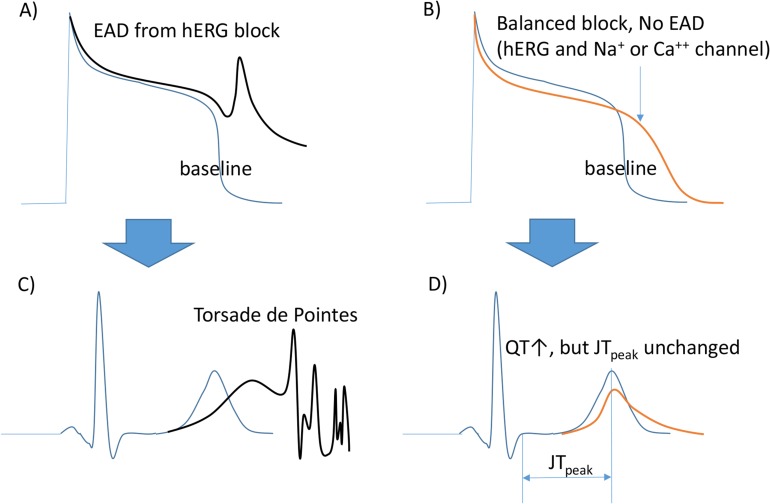 | Figure 2Channels blocked and changes in AP/ECG measures. (A) hERG blockage induced early after depolarization (EAD). (B) Balanced blocking (hERG, the repolarizing current blocker, and Na+ or Ca++ channels, depolarizing current blockers) does not cause EAD. (C) TdP by hERG blocking. (D) The QT is prolonged without changes in the JTpeak length in this ECG. This figure has been adapted from a previous report.[16]
|
Go to :
Four working groups of the CiPA
The CiPA project is composed of four groups. These deal with the in vitro study of ion channels (Group 1), in silico modeling of ion channel study results (Group 2), cardiomyocyte study (Group 3), and biomarker searches for factors other than QTc in human ECGs (Group 4).
In the first year of CiPA project, the members working for Groups 1 and 2 selected 28 drugs with known risks for TdP.[6] These drugs were grouped into high-, intermediate- and low-risk forms based on previous reports and professional opinions. The list is shown in Table 1. Of the 28 drugs, Groups 1 and 2 first selected 12 test drugs to develop appropriate models to simulate APD by modifying the O'Hara-Rudy model[7] and to discover in silico biomarker candidates that might outperform the prolonged APD in predicting the risk of TdP. The performance of the modified model and the proposed biomarkers in the 12 drugs were tested in the other 16 drugs (verification drugs) later.
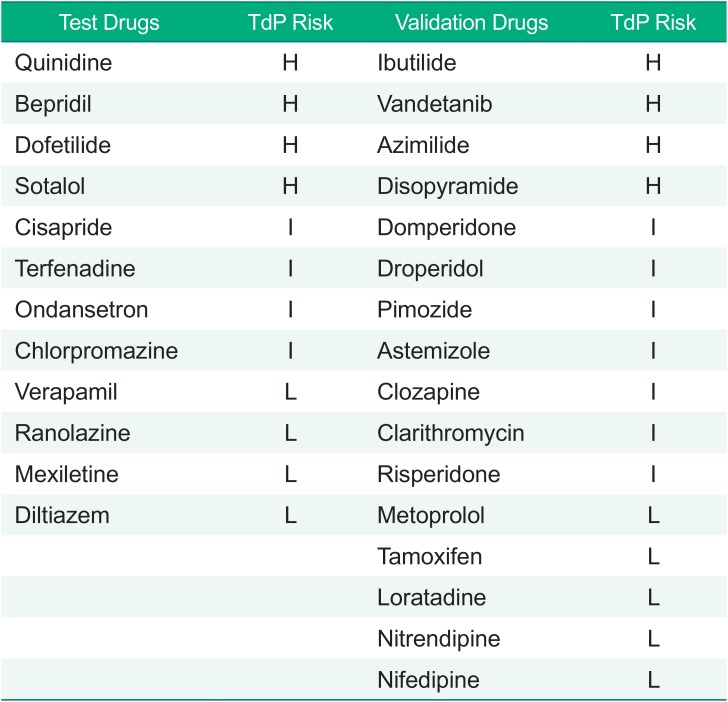
Table 1
Drugs evaluated in the CiPA projects
![]()
Go to :
1. Ion channel working group and 2. In silico modeling group
At the beginning, seven cardiac ion channels in the 12 test drugs—including hERG—were of interest to the CiPA. As for hERG, channel trapping was studied on human embryonic kidney (HEK) cells using Milne's protocol at the FDA.[8] For the other six channels, concentration and channel blocking data for 12 drugs from a previous publication by Crumb at al.[9] were used instead of performing in vitro ion channel studies. With these data, the in silico modeling Group 2 calculated parameters of channel trapping and blocking of hERG, and IC50 and Hill coefficients for the other six channels (Imax was assumed to be 1 in all drugs.). Those values were then used to simulate APs using the modified O'Hara–Rudy model. As uncertainty in the parameters was estimated using a bootstrap method in R and C codes, high performance computing was a necessity. They calculated APD and new markers derived from simulated APs at drug concentrations 1 to 20 times the unbound Cmax. In many of the new markers, qNET—defined as the integral of the net current from the beginning to the end of the simulated beat—was found to perform best in estimating the relative TdP risks between drugs.[10] Therefore, they performed a validation study with the other 16 drugs and the results have been published.[6] At the validation step, the channels studied were hERG, INaF (fast sodium), INaL (slow sodium), and ICaL only, which were the most influential channels on the AP shape. They reported that their modified O'Hara–Rudy model (IKr-dynamic O'Hara–Rudy model) and qNET calculated with the model worked well in discerning the TdP risks of the 16 drugs undergoing validation.
Go to :
Unresolved issues for Groups 1 and 2 of CiPA
The CiPA project is scheduled to be finalized in 2018. Reports published by the ion channel and in silico working groups seem to include promising results. However, there remain some unresolved issues.
One of the goals of developing the in vitro and in silico methods was to apply them to high throughput screening in the drug discovery phase. To be ‘high throughput’, automatized patchclamp methods should be used to measure ion channel blocking instead of the time consuming manual method. However, the huge intra and interlaboratory variability of automatized methods—still unresolved—have been obstacles to such use.[11]
Because it takes well-trained and experienced researchers/technicians to obtain reliable results in manual patch-clamp methods, applications of in vitro/in silico methods of CiPA to high throughput screening in the drug discovery phase are not yet practiced.
The conclusive biomarker proposed by the in vitro and in silico groups of CiPA is qNET. However, the usefulness of this outcome depends on the reliability of the in vitro channel blocking assay results. In the validation study, regardless of the methods (manual or hybrid), they had to use the hERG channel blockage data obtained from a manual method. In the hybrid method, assays for the other channels were done automatically. Thus, a fully automatic method has never been applied to obtain a qNET outcome. If the qNET is to be applied to mass screening during the drug discovery steps, full automatization will be a necessity. Because the ultimate starting point of the qNET metric is the in vitro channel blocking assay result, its reliability is critical. Even when the manual method is used, aberrant results can occur inadvertently, as exemplified in the validation report by the CiPA group.[6] Questions on the necessity of the in silico analysis demanding high-performance computing facilities are still being raised. Mistry has reported that Bnet—a simple metric calculated as the gap in blocking repolarizing (hERG) and depolarizing channels at the unbound Cmax value of each drug—could be used to predict the risk of drugs causing TdP. [12,13]
Go to :
3. Cardiomyocyte working group
This working group has performed AP measurements in human stem cell-derived cardiomyocytes (iPSC-CMs) for the 28 drugs chosen by CiPA.[14] Their methods for measuring AP were indirect, such as microelectrode assays and voltage-sensitive dyes, which are easier than the patch-clamp method. Cardiac-type rhythms and arrhythmic reactions in the iPSC-CMs were observed after applying the test drugs. The merit of observing arrhythmia itself without measuring several channels is that iPSC-CMs have all the cardiac ion channels, as in the normal human heart. An unresolved issue is that the quantitative expression of ion channels in the iPSC-CMs differs from those observed in mature, adult cardiomyocytes. Greater expression of calcium channels in their iPSC-CMs resulted in a shortened APD at a higher concentration of verapamil, the drug blocking both hERG and calcium channels; that outcome is opposite to clinical studies where no QTc shortening was reported. [15] Quantitative or qualitative comparisons of stem cell-derived cardiomyocytes developed by different laboratories using different methods (e.g., human embryonic stem cell-derived cardiomyocytes versus iPSC-CMs, or iPSC-CMs derived from different donors) should be performed to establish their usefulness in reflecting the behavior of mature, adult cardiomyocytes.
Go to :
4. ECG working group
This group has been searching for biomarkers in the ECG other than QTc. Using an analysis of ECG data obtained from FDA-sponsored clinical trials using eight drugs, the heart rate corrected JTpeak interval was proposed as a biomarker. As shown in Figure 2, the working group reported that drugs blocking both the hERG and sodium or calcium channels (depolarizing currents) are expected to increase the QT interval without increasing the JTpeak. Those drugs blocking only hERG (and, thus, increasing the TdP risk at higher Cmax values) should then increase the JTpeak and QTc as their Cmax values increase.
At a workshop held at the Safety Pharmacology Society (SPS) 2018 conference in Washington DC, Vicente presented the ECG analysis results, also summarized in a published article.[16] In his presentation, the ΔΔ JTpeak (placebo and baseline corrected JTpeak) values measured after giving verapamil or ranolazine, balanced channel blockers, were near zero or negative, in contrast with those of hERG-only blockers that were positive or increased according to drug concentrations.
In the hERG-only blockers, whether the ΔΔ JTpeak values increase with the concentration or whether they might simply be greater than some threshold (e.g., 0 ms or 5 ms) as a sign of drug-induced increase in the JTpeak is not yet clearly agreed upon.
Go to :
Conclusion
It is not yet clear whether the CiPA project should conclude in 2018 or be extended further. The new biomarker proposed by CiPA is qNET, in other words, the torsade metric score (mean qNET at 1, 2, 3, or 4 × Cmax). CiPA also proposed using the JTpeak, a new ECG biomarker that might discriminate balanced channel blockers from hERG-only blockers. Although the usefulness of the two new molecular markers needs to be tested in the real world of drug development, they are expected to help the pharmaceutical industry in making right decisions earlier. Furthermore, quantitative validation of the usefulness of stem-cell derived cardiomyocytes on screening for the risk of TdP might facilitate early discovery and development works.
Go to :
 XML Download
XML Download