

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Non-alcoholic fatty liver disease (NAFLD) has emerged as the most prevalent chronic liver disease worldwide, affecting 20–30% of the general population [1]. The identifying NAFLD is hepatic steatosis, which is characterized by fat accumulation in more than 5% of hepatocytes. Fat deposition is also caused by viral infections, certain medications, and genetic disorders [2]. Nonalcoholic steatohepatitis (NASH) is a severe progressed form of NAFLD. The pathological features of NASH include hepatocellular damage and injury, inflammation, and fibrosis and steatosis [3]. The incidence of NASH is calculated to be 2–5% in the general population and is considered as a significant risk factor for hepatic cirrhosis and even hepatocellular carcinoma (HCC).
Although NAFLD and NASH have been studied more than 30 years, it is still not fully elucidated how they are initiated and progressed. Initially, theory for the NASH pathogenesis, two hit hypothesis, was proposed by Day and James [4]. Simple steatosis, such as insulin sensitivity (IR) and excessive fatty acid influx to the liver are two important factors and is the first hit of NASH. Second hit is that excess lipid accumulation in the liver results in oxidative and ER stress, and lipotoxicity, which in turn trigger mitochondrial dysfunction and hepatocyte injury. They can stimulate inflammatory and wound healing responses activate immune cells in liver [2]. The liver has an abundance of macrophages, such as Kupffer cells (KCs) and infiltrated macrophages, compared to other organs. It is estimated there are 20–40 macrophages are supplementing every 100 hepatocytes [5]. They activate hepatic stellate cells, which are the major source of liver fibrosis [6]. As results, activation of inflammatory macrophages and stellate cells promotes the progression from steatosis to NASH [7].
Kupffer cells (KCs) are the resident macrophage in the liver reside in liver sinusoids, the portal tract, and hepatic lymph nodes [8]. In pathologic conditions, bone marrow-derived monocytes, such as infiltrating macrophages (monocyte-derived macrophages, MDM), migrate to the liver and work in collaboration with KCs. However, the difficulty in cellular experimental techniques, the inflammatory responses and function of both KCs and MDMs in the liver are not fully understood on NAFLD and NASH progression.
In this Review, we focus on the discussion about the current knowledge on the origins and composition of hepatic macrophages, including KCs and MDMs, and their involvement in both promoting and resolving liver inflammation, injury, and fibrosis up to NASH.
Origin and composition of hepatic macrophages
Macrophages are a heterogeneous population of immune cells and undertake for innate immune reactions with diverse functions in tissue homeostasis and disease progression and resolution [9]. They recognize, ingest, and degrade cellular debris, foreign material, or pathogens and exert a central function in orchestrating inflammatory processes. However, macrophages employ a wide range of different functions and consist of opposing cellular subsets in the liver [10]. The diverse functions are considered to be plasticity based on their cellular origin and local microenvironment.
The hepatic macrophages have been characterized by two populations; resident and infiltrating macrophages, which are distinguished by ontogeny, phenotype, and functional characterization [7]. Resident liver macrophages, KCs reside in liver-specific and are self-renewing cells which originated from embryonic progenitor cells derived from the yolk sac. They are located in the liver sinusoids in close contact with the liver sinusoidal endothelial cells [11]. They play as first-line guardians, against gut-derived microbiomes, pathogens or foreign bodies. During hepatic damage and injury, they are also assisted by infiltrating macrophages that originate from blood circulating bone marrow-derived monocytes, which call monocyte-derived macrophages (MDMs) [12]. MDMs also play as a pro-inflammatory response, whereas they could convert into alternative or opposing state of activation.
In general, they can be divided either a classical activation (pro-inammatory M1) or an alternative activation (anti-inammatory M2) state by response to different stimuli [13]. M1 is induced by lipopolysaccharides (LPS) or interferon-γ and is associated with the production of proinflammatory cytokines and chemokines. M1-induced cytokines are important for host defense but can induce tissue damage and immune cell activation in the situation of chronic inflammation [14]. The proinflammatory response is reduced by regulatory macrophages (M2), which is induced by interleukin (IL)-4/13, some Toll-like receptor (TLR) signaling pathway, IL-10 or glucocorticoids. M2 produces cytokines and growth factors, concerned in wound healing and recovery from disease. Because they stimulate new vessel and scar formation, M2-derived cytokines and growth factors are essential for tissue remodeling and the pathogenesis of chronic liver disease [13]. The M1 and M2 subtypes represent the different polarization and function states, which can be identified by specific markers, such as M1 markers is peroxisome proliferator-activated receptor (PPAR), CCL2, CX3CL1 and CCL5 whereas M2 markers is IL-4 receptor (IL-4R), mannose receptor (MR/CD206), and arginase1 (Arg-1) [1516].
These studies suggest that disruption of the M1/M2 balance could be effect on development, homeostasis and diseases and regulation of polarization of macrophage populations toward different phenotypes has been implicated in clinical application. In addition, activation and polarization of macrophages are deeply implicated in the pathogenesis and progress in chronic liver diseases, such as NAFLD and NASH.
Kupffer cells (KCs)
The liver is comprised by the hepatocyte, the most numerous 60% of the total liver cells and 80% of the volume of liver [17]. Twenty % of the liver cells include sinusoidal endothelial cells KCs and hepatic stellate cells. Approximately 15% of liver cells are KCs, the resident macrophages of the liver, are the largest tissue specific population of macrophages in the body [18]. KCs are located in liver sinusoids, the portal tract and hepatic lymph nodes, which are the crossroads of the portal vein and hepatic artery tributaries.
The main function of KCs in healthy liver is the phagocytosis and presentation of pathogens from portal vein and arterial circulation, representing one of the first lines of defense of the organism. Under normal conditions, KCs express low levels of MHC (Major Histocompatibility Complex) class II and co-stimulatory molecules and then inhibit T cell activation [19]. KCs also participate in immunosuppression by expressing high amounts of T cell suppression molecules such as IL-10 and TGF-β and low levels of costimulatory molecules, which is critical for the induction of tolerance to hepatocyte-expressed antigens [202122]. Moreover, KCs remove microbes, suggesting KCs are preserving the hepatic area at optimally regulated stage of inflammation [8]. For example, KC surface exhibits TLRs, which are responsive to LPS, and produce and secrete anti-inflammatory signals to respond to lipotoxicity, including IL-10 in response to LPS [23].
The circumstance of NAFLD to NASH, various metabolic syndromes and insulin resistance, could induce accumulation of free fatty acids and lipids in peripheral blood and hepatocytes, and could stimulate innate immune responses that result from the stimulation of lipotoxins and LPS [24]. KCs play a critical mediator in the development of NAFLD. One of the signaling pathways that appear to be involved in this disorder is LPS/TLR4. Binding of LPS to TLR4 on the KC surface activates NF-κB pathway and increases the production and secretion of pro-inflammatory cytokines, which recruit lymphocytes and other leukocytes [25]. It is also reported that high accumulation of lipids in KCs induces mitochondrial dysfunction and endoplasmic reticulum (ER) stress [2627]. ER stress promotes activation of the NF-κB, JNK and CEBP pathway in KCs, and results in insulin resistance and apoptosis.
These data suggest that KCs play a critical role in maintain liver functions and activation of Kupffer cells will determine their functions in liver damage and metabolic disorder.
Recruited monocyte-derived macrophages (MDMs)
Previously, research on samples from patients and mouse with liver diseases has clarified the heterogeneity of macrophages in the liver. They distinguished the tissue-resident macrophages (KCs) from infiltrating macrophages called monocyte-derived macrophages (MDMs) [10]. KCs and MDMs can be discriminated by the origin, the differentiation, the behavior and, most importantly, the functions on homeostasis and disease. It has been known that KCs are established during embryonic development from the fetal yolk-sack [28], whereas MDMs originate from circulating adult blood monocytes. Their function is also extraordinarily versatile, for example, KCs respond to environmental signals from liver tissue or blood circulation in a highly diversified manner. But, MDMs can rapidly develop into restorative, tissue-repairing cells in diseases environments.
As a macrophage maker in human liver, CD68 has been proposed but non-distinct marker for Kupffer cells [29]. Recently, CD163L was proposed as a marker for KCs, in contrast CLEC5A is identified pro-inflammatory MDMs [30]. In mice, Ly-6C (Gr-1) and CCR2 expression levels is high in circulating monocytes, the precursors for macrophages and dendritic cells, and they rapidly infiltrated in tissue upon liver injury [31]. The human counterparts of these monocyte subsets are termed classical (CD14, CD162) [32]. Upon liver damage or injury, Ly-6Chi MDMs are hugely recruited to the liver and prevail the macrophage pool in mice [33]. Although these monocyte-derived macrophages initially play pro-inflammatory and anti-inflammatory actions, they adjust for tissue repair and injury resolution. The immerse infiltration of MDMs is a characteristic feature of liver damage or injury in humans [34].
Recent studies have been distinguished the hepatic macrophages by various cell surface markers that infiltrated MDMs play the critical role in orchestrating liver fibrosis progression. However, different subsets of infiltrated MDMs play opposite roles in liver fibrosis. In the early phase of tissue damage or injury, increment of CCL2 expression recruits CCR2hi/Ly-6Chi monocytes in liver tissue [35]. The infiltrated Ly-6Chi monocytes differentiate into pro-inflammatory macrophages, and they directly interact with hepatic stellate cells (HSCs). HSCs produces pro-fibrotic cytokine, TGFβ and accelerate hepatic fibrosis [6]. It suggests a pro-fibrotic function of the Ly-6Chi hepatic macrophages. In contrast, increased phagocytosis of macrophages assists the phenotypic switch of the subsets, Ly-6Chi macrophages to Ly-6Clow macrophages [36]. The Ly-6Clow macrophages produce Metrix Metalloproteases (MMPs) and orchestrates the resolution of liver fibrosis [37]. The finding supports for the anti-fibrotic function of the hepatic macrophages. These data propose that infiltrated MDMs play a critical role in liver homeostasis and diseases and need to be further study.
Involvement of hepatic macrophages in progression of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis
The hallmark of NAFLD is an excess of fatty acids and accumulation of lipids in the liver. They result in lipotoxicity to induce liver damage and injury [2538]. These lipid-induced liver damage signals are the initiator for dramatic expansion of hepatic macrophages because of the massive influx of monocytes into the liver. It was reported that cholesterol crystals in the livers progress both human and mouse NASH [39]. KCs take up cholesterol-rich lipoproteins using scavenger receptors [40]. Lipid accumulated KCs recruit CD4+ T lymphocytes and increase T cell tolerance [19]. Cholesterol crystals also activate NLR proteins (NLRP) in LPS-exposed KCs [41]. Recently, low-density lipoprotein cholesterol formed cholesterol crystals on the lipid droplet membrane of hepatocytes and activated THP1 macrophage cells that upregulated TNF-α, NLRP3, and IL-1β mRNA [42]. It suggested that reduction of cholesterol levels result in resolution of NASH through dissolution of cholesterol crystals and disperse KC structures.
In addition to lipotoxicity, hepatocyte cell death is a critical process in NAFLD and NASH. Death receptors, such as TNF receptors, FAS, and TRAIL receptors, have been shown to mediate inflammatory signaling [4344]. Cells undergoing necrosis and apoptosis release damage associated molecular pattern (DAMP) [45], which further accelerate inflammation by activating inflammasomes [46]. The activation of NLRP cause the assembly of the inflammasome complex, which contains caspase-1, propagating inflammation and cell death by cleavage of prointerleukins into their interleukin form, such as IL-1β, IL-18 [4748]. The inflammasomes are a critical in the progression to NASH, leading to fibrosis and cirrhosis. The DAMP associated with M1 macrophages secrete high motility group box 1 (HMGB1), heat shock proteins and also breakdown products of extracellular matrix. Apoptosis is increased in hepatic cells as a result of excess lipid-uptake [49]. Saturated fatty acids induces hepatocyte damage by multiple mechanisms. Excess accumulation of lipids cause lipotoxicity, which occur lipotoxic stress in the endoplasmic reticulum and mitochondria, thereby leading apoptosis induction of hepatocytes [5051]. ER stress related proteins, such as IRE1, PERK, and ATF6 upregulate p53, which induces the expression of BAX and the release of cytochrome C from mitochondria result in apoptotic death [52].
In the progression of deteriorating conditions from NAFLD to NASH, cells release stress signals. Especially, hepatocytes undergoing necrosis release DAMP and chemo-attractants and recruit and activates various immune cells to the liver, initiating a wound healing & repair responses by fibroinflammatory reactions [45]. This inflammatory response can boost to lead fibrosis and cirrhosis. In this process, several cytokines and chemokines, such as TNF-α, IL-1β, IL-12, IL-23, IL-6, CCL2, and CCL5 are released in liver [35]. The accumulation of these cytokines in liver accelerate further the release of DAMP, promoting further hepatocyte damage, activating and recruiting leukocytes to further increase the inflammatory response in the fibroinflammatory process.
Resident KCs are important in the response by rapid producing cytokines and chemokines, such as tumor necrosis factor (TNF)α, IL-1β, CCL2, and CCL5, which promote the recruitment of other immune cells, such as monocytes [3553]. For example, in mouse models of CCL4-induced liver fibrosis, the infiltrated monocytes are dominated by the Ly-6Chi subset, after transmigration into the liver, the Ly-6Chi monocytes differentiate into Ly-6Chi macrophages. Other study suggested that the tissue micro-environmental factors during NAFLD process could promote the switch of Ly-6Chi macrophages into Ly-6Chi macrophages [36]. It is important to target hepatic macrophages for hepatic disease therapy, we have to understanding the respective role of hepatic macrophages, resident KCS and infiltrated Ly-6Chi and Ly-6Clow macrophages in various types of liver diseases. KC are activated by CD14, when presence of LPS activates TLR4 signaling pathway. It was reported that high expression of CD14 increases the sensitivity to LPS in cells [54]. KCs are also activated by NF-κB, MAPK, ERK1, p38, JNK, and IRF3 signaling pathway [55]. In NAFLD and NASH, a high expression of TLR4 results in massive release of cytokines, and thus contributes to progression of inflammation and fibrosis [56].
The recruitment of Ly-6Chi bone marrow-derived monocytes has been shown to be a critical event for the promotion of NAFLD and NASH. The chemokine interactions, such as CCL1-CCR8, CCL2-CCR2, CCL5-CCR5, and CXCL10-CXCR3, have been shown to recruit monocytes as well. These bone marrow-derived monocytes collaborate with KCs to promote inflammation, inducing chemokines and cytokines such as TNF-α and IL-1β [81034].
Chemokine receptor CXCR3, a CXCL10 receptor, mediates bone marrow-derived monocytes infiltration and secretion of inflammatory cytokines [57]. CCL2 and CCL5 from macrophages activate HSCs leading to fibrosis. TNF-α has been shown to be a crucial factor for NASH by promoting bone marrow-derived monocytes infiltration. TNF-α can activate apoptosis and necroptosis through death receptors, and also NF-κB pathway to promote inflammasome, thereby accelerating inflammation. TNF-α alienates adiponectin, an anti-inflammatory adipokine, and then induces inflammation and insulin resistance [58]. The increment of TNF-α has a key role in the progression of NASH by promoting this inflammation.
Neutrophils have presented an important role in activation of hepatic macrophages and NASH progression [59]. Activation of TLR4 signaling on hepatic macrophages increases neutrophil adhesion in liver sinusoids [60]. Neutrophils also promotes further recruitment of macrophages using an antigen presenting method. Additionally, neutrophils accelerate fibrosis by activation and increment of proliferation of HSCs [6162].
Conclusion
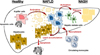
It has been become distinct that hepatic macrophages are a critical to initiating and propagating hepatic inflammation to progress NDFLD and NASH and is summarized in Figure 1. Targeting hepatic macrophages seems to be a promising therapeutic approach to care liver diseases, such as NAFLD and NASH. Now, many therapeutic options and trial are developing and discovering to treat NAFLD and NASH by targeting macrophages.
However, there are still need to be overcome in targeting human liver macrophages. Firstly, we have to distinguish the differences between mouse models and humans, although there is substantial similarity. Experimental conditions of mice are not the same as human diseases. Moreover, many of the therapies improve fibrosis, but not fully overwhelm NASH. Therefore, although the clinical implications of fibrosis resolution are very beneficial, the need to find a more encompassing treatment remains. There remains many points in the molecular mechanisms of NAFLD and NASH that can be deliberated and explored for their therapeutic potentials. Targeting macrophages to threat NAFLD and NASH in its early progress stages may be a superior method to reduce the damage, compared to do it on later stages of NASH and cirrhosis.
 XML Download
XML Download