

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is the most common adult-onset motor neuron disease, and is characterized by progressive muscle atrophy and weakness that eventually results in respiratory failure. Sporadic ALS (sALS) comprises more than 90% of all ALS cases. Although more than 30 ALS-associated genes have been reported, the mechanism of motor neuronal degeneration remains unclear. Nuclear pore complexes (NPCs) are central to the process of nucleocytoplasmic transport, and they are scattered over the nuclear membrane. Each NPC is assembled from approximately 30 evolutionarily conserved proteins known as nucleoporins.1 Recent studies have found that the disruption of nucleocytoplasmic transport through the NPC is a potential cause of neuronal death in C9orf72 mouse models of ALS.234 Moreover, morphological changes in the nuclear membrane are associated with the disruption of nucleoporins in the motor neurons of patients with sALS or SOD1-associated ALS.567 In addition, mutations in the gene encoding GLE1 (a nucleoporin) have been reported in patients with ALS.8
Disruption of nucleoporins has been reported in an sALS mouse model (ADAR2flox/flox/VAChT-Cre or AR2 mice),9 in which the adenosine deaminase acting on RNA 2 (ADAR2) was conditionally knocked out.10 Analogous changes in nucleoporins have also been observed in preliminary studies of the ADAR2-deficient motor neurons of sALS patients.10 ADAR2 regulates Ca2+ influx through α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors via adenosine-to-inosine conversion at the glutamine/arginine site of GluA2 mRNA, which makes ADAR2 is a key factor in acquired Ca2+ resistance in motor neurons. Disease-specific and site-selective deficiency of ADAR2 has been demonstrated in sALS motor neurons,111213 and ADAR2 deficiency is closely associated with the death of motor neurons in conditional ADAR2-knockout mice.9 Notably, ADAR2 down-regulation along with pathology of the 43-kDa TAR DNA-binding protein (TDP-43) that is one of the most-reliable pathological hallmarks of ALS1415 have been observed concomitantly in the motor neurons of ALS patients.16 The mechanism underlying the co-occurrence of these two molecular abnormalities within the same neurons involves an exaggerated Ca2+ influx and activation of calpain (a Ca2+-dependent cysteine protease) in the ADAR2-deficient motor neurons, and the resultant calpain-dependent TDP-43 fragments serve as seeds for TDP-43 aggregation.17 These findings suggest that ADAR2 deficiency is involved in the pathophysiology of motor neuron degeneration in sALS.
TDP-43 pathology is invariably observed in motor neurons that are devoid of ADAR2.16 We suspect that the precise morphological relationship between the formation of TDP-43 aggregation and the alteration of nucleoporin expression in sALS motor neurons further reflects the role of nucleoporins in ALS. In this study we examined the expression of nucleoporin p62 (NUP62), a central nucleoporin containing phenylalanine-glycine repeats, and karyopherin beta 1 (KPNB1), a nucleocytoplasmic transport master regulatory protein,18 in relation to TDP-43 pathology.
METHODS
Human subjects
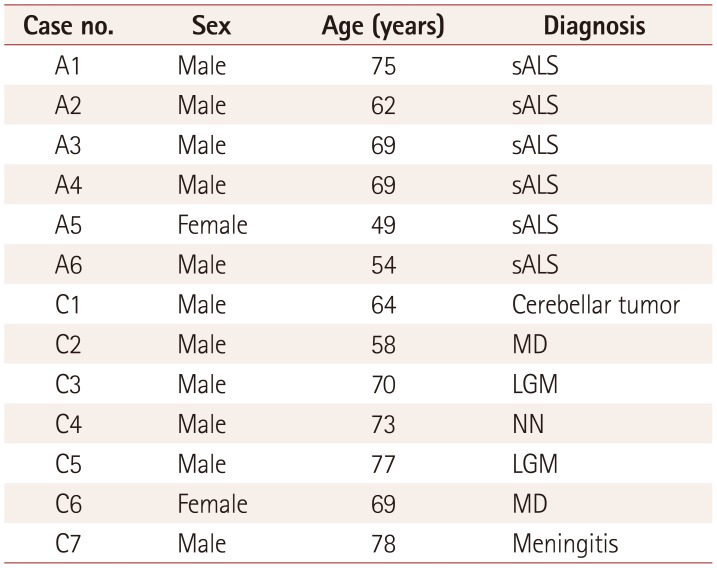
Spinal cords were obtained from seven control cases and six sALS patients after autopsy. The cases used in this study are listed in Table 1. Written informed consent for performing the autopsy and approval to use autopsy tissue specimens for research purposes was obtained from each patient or their family. The research protocol used in this study was approved by the Institutional Human Ethics Committee of Tokyo Medical University (No. 3374).
Immunofluorescence analysis
Upper-, middle-, and lower-level lumbar spinal-cord sections were obtained from human ALS patients and controls, then fixed in 10% neutral buffered formalin for approximately 7 days. They were then embedded in paraffin, and 4-µm-thick sections were deparaffinized in xylene and hydrated in a graded series of ethanol solutions. The sections were heated in citrate buffer (pH 6.0) at 120℃ for 10 min to allow antigen retrieval, then washed with phosphate-buffered saline (PBS) and incubated with a primary antibody overnight at 4℃. The sections were then washed with PBS, and incubated with a secondary antibody at room temperature for 1 h.
The following primary antibodies were used: rabbit polyclonal and mouse anti-TDP-43 (1:3,000 dilution of 10782-2-AP, Proteintech Group Inc., Rosemont, IL, USA; and 1:3,000 dilution of H00023435-M01, Abnova Co., Taipei, Taiwan), mouse anti-NUP62 (1:200 610497, BD Biosciences Inc., Franklin Lakes, NY, USA) and rabbit anti-KPNB1 (1:200 sc-11367, Santa Cruz Biotechnology Inc., Dallas, TX, USA). The following species-appropriate secondary antibodies were used: Alexa Fluor 488, 594, or 633 secondary antibodies at 1:1,000 dilution (Invitrogen Inc, Waltham, MA, USA). The sections were stained with DAPI (D1306, Molecular Probes Inc., Eugene, OR, USA) for 5 min at room temperature for nuclear staining before being mounted onto slides with an antifade reagent (Fluoromount/Plus K048, Diagnostic Biosystems Inc., Pleasanton, CA, USA). The sections were then examined under a fluorescence microscope (BIOZERO BZ-8100, Keyence Corp., Osaka, Japan).
Three levels of lumbar sections from each subject were used to quantify TDP-43 and NUP62 immunoreactivity in the motor neurons. On each section, neurons with nucleoli in the anterior horns were counted separately. Statistical analyses (t-tests) were performed with IBM SPSS Statistics software (version 24; IBM Corp., Armonk, NY, USA). Data presented are mean±SD values.
RESULTS
Detection of TDP-43 and NPC proteins by immunofluorescence staining
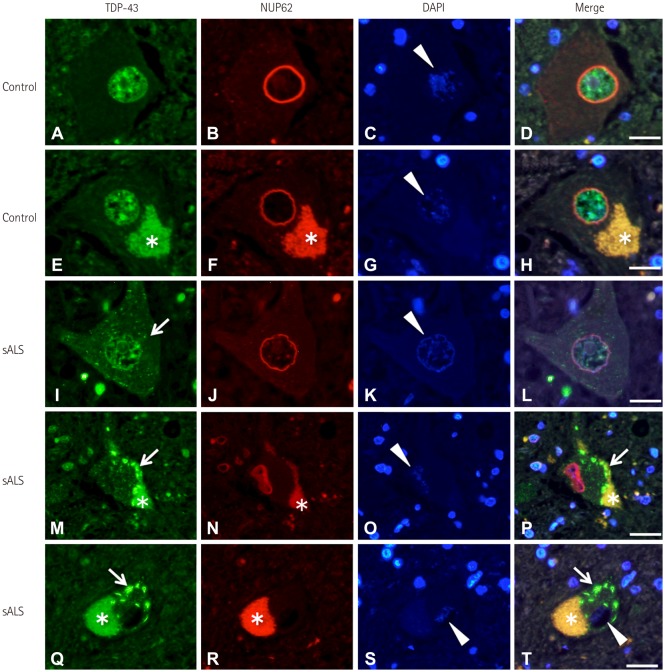
Immunofluorescence staining revealed that in the normal spinal motor neurons, TDP-43 was localized in the nucleus while NUP62 immunoreactivity appeared as a smooth round rim bordering the nuclear margin (Fig. 1A–H). In the sALS spinal anterior horn, some motor neurons exhibited decreased nuclear TDP-43 immunoreactivity with fine cytoplasmic TDP-43-positive inclusions (Fig. 1I); these neurons also demonstrated circular NUP62 immunoreactivity (Fig. 1J). Spinal motor neurons that had apparent TDP-43 mislocalization showed irregular, disrupted nuclear staining of NUP62 (Fig. 1M–P). Some atrophic spinal motor neurons that showed TDP-43 mislocalization demonstrated no NUP62 immunoreactivity (Fig. 1Q–T).
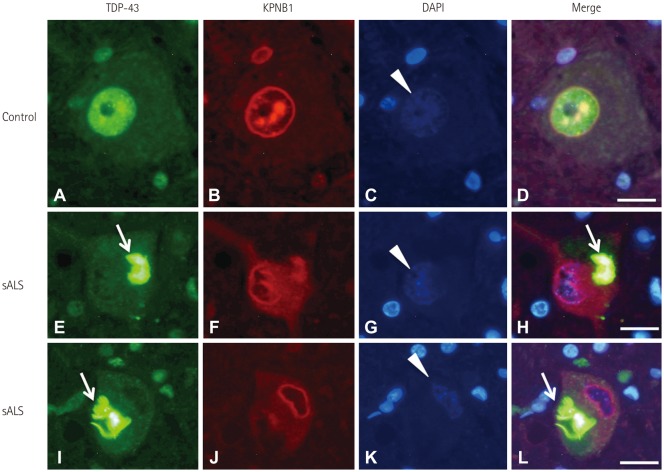
KPNB1 immunoreactivity was observed in the nuclear margin as a distinct round circle or dots in the nucleus of normal motor neurons (Fig. 2A–D), whereas KPNB1 immunoreactivity was observed as an irregularly twisted line in the disrupted or deformed nucleus, and was diffusely expressed within the cytoplasm of neurons with mislocalized TDP-43 (Fig. 2E–L). These observations suggest that mislocalization of TDP-43 is correlated with disruption of nuclear staining by anti-NUP62 and anti-KPNB1 antibodies.
Quantification of TDP-43 and NUP62 immunoreactivity in the motor neurons
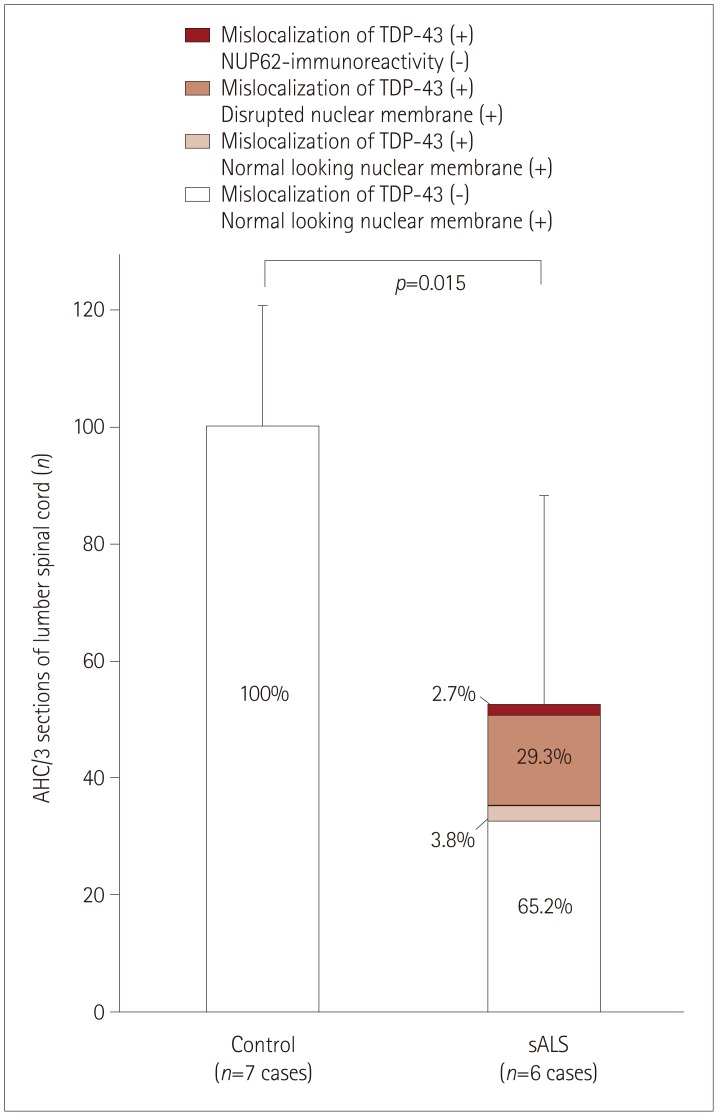
Anterior horn cells (AHCs) with nucleoli on three levels of lumbar sections were counted separately. There were fewer AHCs in sALS patients (52.2±36.4 cells) than in control subjects (104.6±15.8 cells) (p=0.015). All lumbar spinal motor neurons from control subjects exhibited normal nuclear TDP-43 immunoreactivity and normal ring-like NUP62 nuclear staining. Among the sALS samples, 65.2% of neurons showed normal nuclear TDP-43 immunoreactivity and normal nuclear NUP62 staining, while the remaining 34.8% exhibited TDP-43 mislocalization (Fig. 3): 3.8% exhibited normal nuclear NUP62 immunoreactivity, 29.3% demonstrated disrupted NUP62 nuclear staining, and 2.7% were negative for NUP62 immunoreactivity. The neurons with disrupted NUP62 nuclear staining or that were negative for NUP62 immunoreactivity were atrophic (Fig. 1M–T).
DISCUSSION
This study found that disruption of the NPC was correlated with TDP-43 mislocalization in the spinal motor neurons of patients with sALS. TDP-43 pathology occurs in motor neurons that demonstrate abnormal activation of calpains, and so we suspect that disruption of the NPC in sALS is related to calpain activation.10 NUP62 immunoreactivity was observed along the round-shaped nuclear membrane in normal motor neurons (Fig. 1A–H). Neurons that exhibited reduced nuclear TDP-43 immunoreactivity with fine cytoplasmic TDP-43-positive inclusions demonstrated circular NUP62 immunoreactivity (Fig. 1I–L), suggesting that these motor neurons are in the early stages of degeneration. The pattern of NUP62 immunoreactivity was irregular within the nucleus of motor neurons that exhibited TDP-43 mislocalization (Fig. 1M–P). Some atrophic spinal motor neurons with TDP-43 mislocalization demonstrated no NUP62 immunoreactivity (Fig. 1Q–T), which is indicative of the end stages of degeneration. The observed changes in NUP62 immunoreactivity seemed to be closely related to the stage of TDP-43 pathology. Similar to NUP62, the distribution of KPNB1 (a molecule involved in nucleocytoplasmic transport) was normal in motor neurons with correctly localized nuclear TDP-43, but was disrupted in motor neurons that exhibited TDP-43 mislocalization.
Activation of the Ca2+-dependent cysteine protease calpain specifically cleaves TDP-43 at the C-terminal region to generate aggregation-prone N-terminal fragments.17 ADAR2 deficiency induces the activation of calpain via the abnormal expression of Ca2+-permeable AMPA receptors, thereby resulting in the abnormal mislocalization of TDP-43 in conditional ADAR2-knockout mice and sALS patients.1719 A particularly interesting observation was that nucleoporins were also cleaved by calpain, and were disrupted in ADAR2-negative neurons that exhibited positive staining for 136kf (a marker for activated calpain) in the conditional ADAR2-knockout mice.17 In the present study, only a portion of the motor neurons with TDP-43 mislocalization exhibited normal nucleoporin staining, whereas all motor neurons with normal TDP-43 immunoreactivity exhibited normal nucleoporin staining. Thus, the calpain-dependent cleavage of TDP-43 in the cytoplasm may precede the cleavage of nucleoporins, as a result of a difference in subcellular localization or of a vulnerability to calpain cleavage between TDP-43 and nucleoporins. These findings suggest that calpain-dependent nucleoporin disruption participates in sALS pathogenesis.17 The mechanism underlying how TDP-43 pathology induces cell death has not been resolved; however, disruption of the NPC induces cell death by inhibiting gene expression that is necessary for cell survival,17 which has been reported to cause motor neuron death.20
Irregular nuclear membrane staining with nucleoporins and abnormal nuclear transporter proteins has been noted in sALS and SOD1-associated ALS, suggesting that nucleocytoplasmic transport is dysfunctional in ALS.4 Morphological abnormalities and disrupted nucleocytoplasmic transport have recently been reported in both in vitro and in vivo models of C9orf72-related ALS234 and sALS.7 Combining these findings with those of the present study suggests that the disruption of nucleocytoplasmic transport underlies the pathomechanism of motor neuronal degeneration in sALS, as well as in some cases of genetic ALS. Further studies are required to elucidate the role of the NPC in nucleocytoplasmic transport in sALS.
 XML Download
XML Download