

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Dear Editor,
A 45-year-old male presented with progressive gait impairment that had first appeared several years previously. He did not complain of any additional symptoms, and denied suffering from any previous medical illnesses. In the neurological examination he showed spasticity, hyperreflexia, positive Babinski signs in the lower extremities, and bilateral ankle clonus. He reported that his mother was the only family member to show a similar gait difficulty, and our assessment of her revealed the same corticospinal tract signs in the lower limbs. Because a positive family history, normal brain MRI, and normal spine MRI were observed, and the findings of serological tests related to spastic paraparesis were all negative, we could only tentatively diagnose hereditary spastic paraplegia (HSP). We therefore performed a genetic study of the patient and his mother.
Molecular genetic studies revealed that both the patient and his mother had a heterozygous c.1413+4A>G (IVS11+4A>G) variant in SPAST, and the Human Splicing Finder tool (http://www.umd.be/HSF3/) predicted that this variant broke the natural donor splice site. This mutation was found neither in the public version of The Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php) nor two population databases: gnomAD (http://gnomad.broadinstitute.org/) and Korean Reference Genome DB (KRGDB; http://152.99.75.168/KRGDB/menuPages/firstInfo.jsp).
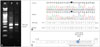
To investigate the effect of c.1413+4A>G on splicing, we performed a reverse-transcription polymerase chain reaction (RT-PCR) using the following primer pair spanning exons 10 to 14 of SPAST: 5′-ACGTGGGAGAAGGAGAGAAAT-3′ (forward) and 5′-TTTGGGTCAATGGACTTCCT-3′ (reverse). The RT-PCR revealed an aberrant splicing product, and the skipping of SPAST exon 11 [r.1322_1413del92 (p.Asp441Glyfs*7)] was confirmed by direct sequencing analysis (Fig. 1A and B). According to 2015 American College of Medical Genetics and Genomics standards and guidelines for the interpretation of sequence variants, c.1413+4A>G was classified as likely pathogenic, since 1 strong (splicing defect confirmed by RT-PCR) and 1 moderate evidence (absence from controls) were satisfied.1
HSP is a genetically heterogeneous disease and its mutations have been identified in more than 70 loci (SPG1–72) and 50 genes.2 Among them, mutations in the SPG4/SPAST gene are the most common cause of HSP, accounting for 15–40% of all HSP cases. SPAST contains 17 exons and encodes the enzyme spastin, which is a member of the ATPase associated with various cellular activities (AAA) protein family.3 Various mutations have been found in all locations in SPAST (nonsense, splicing, and small indels) or mainly clustered in the AAA domain (missense indels). These mutations could impair spastin function through either a loss-of-function or a dominant-negative effect, and may lead to a disturbance in the formation of the microtubule cytoskeleton. This is a plausible pathomechanism of HSP.34 An obvious genotype–phenotype correlation has not been elucidated previously. Considerable phenotypic variability (age at onset, rate of progression, and degree of disability) has been observed both between and within individual families, even for the same HSP gene mutation.5 Instead, longer disease duration or the presence of intragenic modifier alleles seem to make the symptoms more severe and produce an earlier age at onset. 67 Previous reports have generally described HSP patients carrying IVS11+1, +2, +3, +5, and +6 splice-site variants as exhibiting clinically pure-type HSP (Supplement Table 1 in the online-only Data Supplement).
In the present patient we discovered a novel likely pathogenic variant of SPAST causing exon-11 skipping, which may affect the function of the AAA domain (Fig. 1C).
 XML Download
XML Download