

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Bone is a metabolically dynamic tissue that is continuously remodeled and renewed. Bone remodeling involves osteoclast-mediated bone resorption followed by osteoblast-mediated bone formation [1], and the precise balance between these two processes is indispensable to guarantee the maintenance of a healthy bone structure in adults [2]. An imbalance in bone remodeling can adversely affect bone structure and function, resulting in skeletal abnormalities such as osteoporosis or osteopetrosis [3].
Osteoclasts are giant multinucleated cells originating from the hematopoietic monocyte/macrophage precursors through osteoclastogenesis. Osteoclastogenesis can be sequentially divided into differentiation of hematopoietic precursors to tartrate-resistant acid phosphatase (TRAP)-positive mononucleated cells or preosteoclasts, followed by formation of multinucleated osteoclasts, and eventual osteoclast activation for bone resorption [4]. In the bone microenvironment, a number of essential molecules for osteoclastogenesis such as tumor necrosis factor (TNF)-related cytokines, receptor activator of nuclear factor-κB ligand (RANKL), polypeptide growth factor, and macrophage-colony stimulating factor (M-CSF) have been reported [5]. M-CSF stimulates receptor activator of nuclear factor-κB (RANK) expression through binding to C-FMS (the M-CSF receptor). RANKL is expressed in osteoblasts either in a membrane-bound or soluble form [6]. The binding of RANK with its ligand, RANKL is believed to be pivotal for the initiation of osteoclast differentiation and subsequent activation of osteoclastic bone resorption [7].
Propofol has been widely used as an intravenous anesthetic agent for general anesthesia and procedural sedation. The molecular structure of propofol consists of a phenolic hydroxyl group and is similar to α-tocopherol or vitamin E [8]. In a recent study, α-tocopherol treatment was reported to increase the number of TRAP-positive multinucleated osteoclasts and the percentage of large osteoclasts by inducing osteoclast fusion via upregulation of dendritic cell-specific transmembrane protein (DC-STAMP) [9]. DC-STAMP is a protein with a seven-transmembrane domain, which is known to be expressed in dendritic cells and IL-4-stimulated macrophages. DC-STAMP is indispensable for cell fusion of osteoclasts during osteoclastogenesis [101112]. This study, therefore, addresses the hypothesis that propofol might positively regulate osteoclastogenesis and bone resorption, and investigates the signaling pathways involved in osteoclastogenic gene expression using bone marrow-derived macrophages (BMMs).
Go to : 
MATERIALS AND METHODS
1. Reagents
Propofol was purchased from Fresenius Kabi Austria GmbH (Hafnerstrabe, Austria), and was freshly prepared as a solution in dimethyl sulfoxide for each experiment. Recombinant soluble RANKL and M-CSF were purchased from PeproTech (Rocky Hill, NJ, USA). Dentin discs were obtained from Immunodiagnostics Systems (Scottsdale, AZ, USA). The Leukocyte Acid Phosphatase (TRAP) kit and anti-β-actin antibody were purchased from Sigma-Aldrich (St. Louis, MO, USA). Antibodies against c-Fos and nuclear factor of activated T cell cytoplasmic 1 (NFATc1) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against p38 and phospho-p38 were obtained from Cell Signaling Technology (Danvers, MA, USA). SB203580, a selective inhibitor of p38, was purchased from Calbiochem (San Diego, CA, USA).
2. Isolation of BMMs
We used BMMs obtained from 5-week-old female ICR mice as osteoclast precursor cells. In brief, whole bone marrow cells were flushed out from the tibiae and femurs of mice with α-modified Eagle medium (α-MEM, WelGENE Inc, Daegu, Korea). After removing red blood cells with a hypotonic ammonium-chloride-potassium buffer and discarding adherent cells, the floating cells were further cultured in the presence of M-CSF (30 ng/ml). M-CSF was always included in the culture medium to guarantee survival of macrophages except in the case of starvation. After 3 days, the cells adherent in culture and were considered as osteoclast precursor cells (BMMs).
3. Cell viability and proliferation assays
Cell viability and proliferation were determined by a colorimetric method using 3-(4,5-dimethylthiazol)-2,5-diphenyltetrazolium bromide (MTT) assay (Sigma-Aldrich). BMMs were seeded in 96-well plates and treated with 0–100 µM propofol for up to 3 days. The cells were exposed to 0, 5, 10, 20, 50, and 100 µM of propofol to determine the concentration of propofol for subsequent experiments. For proliferation assays, cells were further incubated with fresh medium, containing 0.5 mg/ml MTT solution for 4 hours. After incubation, blue formazan product formation was measured with an enzyme-linked immunosorbent assay plate reader at a wavelength of 570 nm.
4. In vitro osteoclastogenesis and TRAP staining
In vitro osteoclastogenesis was induced by culturing BMMs in the osteoclastogenic medium (α-MEM containing 30 ng/ml M-CSF and 100 ng/ml RANKL) for 4 days. Fully mature osteoclasts were formed between day 4 and 5. Mature osteoclasts were visualized by TRAP staining and TRAP-positive cells were counted using an Olympus 23 light microscope (Olympus, Tokyo, Japan). TRAP-positive multinucleated (≥4 nuclei) cells were counted as osteoclasts.
5. Bone resorption assay
For the in vitro bone resorption assay, BMMs were plated on dentin discs containing mineralized collagen matrix and were allowed to differentiate into osteoclasts using various concentrations of propofol (0–50 µM) for 5 days. After removing adherent cells with extensive washing, dentin discs containing 2 × 103 cells were observed under a laser-scanning confocal microscope (Zeiss LSM 5 PASCAL). The resorbed area was measured through the Zeiss LSM Image Browser software (version 3.0).
6. Western blotting
Western blotting was performed following our standard protocol. Specifically, whole cell lysates were prepared by lysing cultured cells in RIPA buffer (50 mM Tris; pH 8.0, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 1% Triton X-100, 10 mM NaF and complete protease inhibitor cocktail). Protein lysates (30–45 µg) were electrophoresed on a 10% gel using SDS-PAGE. Following protein transfer, nitrocellulose membranes were blocked with 5% skimmed milk for 1 h at room temperature. Immunoblotting was performed with polyclonal antibodies specific to c-Fos, NFATc1, p38, and phospho-p38. After incubation with HRP-conjugated secondary antibodies for 1 h, the immunoreactivity of membranes was detected through chemiluminescence using an ECL detection kit (Amersham Biosciences, Buckinghamshire, United Kingdom). The equal loading of lysates was verified using β-actin.
7. Real-time PCR and RT-PCR
RNA extraction was performed using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA), and 2 µg of RNA was reverse-transcribed using Superscript II (Invitrogen) according to the manufacturer's instructions. For quantitative PCR analysis, cDNAs were amplified using SYBR green PCR master mix (Applied Biosystems, Warrington, Cheshire, UK) in an AB7500 Real-Time PCR system (Applied Biosystems) for 40 cycles of 15 s denaturation at 95℃ and 1 min amplification at 60℃. The DC-STAMP mRNA levels were normalized against actin mRNA levels. The primer sequences used in PCR analysis were as follows: c-Fos, 5′-CTGGTGCAGCCCACTCTGGTC-3′ (forward) and 5′-CTTTCAGCAGATTGGCAATGCC-3′(reverse); NFATc1, 5′-CAACGCCCTGACCACCGATAG-3′ (forward) and 5′-GGCTGCCTTCCGTCTCAT-AGT-3′ (reverse); TRAP, 5′-ACTTCCCCAGCCCTTACTACCG-3′ (forward) and 5′-TCAGCACATAGCCCA-CACCG-3′ (reverse); DC-STAMP, 5′-GACCTTGGGCACCAGTATTT-3′ (forward) and 5′-CAAAGCAACA-GACTCCCAAA-3′ (reverse); and Actin, 5′-GTGGGGCGCCCCAGGCACCA-3′ (forward) and 5′-CTCCTTAAT-GTCACGCACGATTTC-3′ (reverse).
8. Statistical analysis
Results are presented as mean ± standard deviation (SD) of three independent experiments. To analyze statistical significance, student's t-test was used and P < 0.05 was considered significant.
Go to :
RESULTS
1. Effect of propofol on cell viability
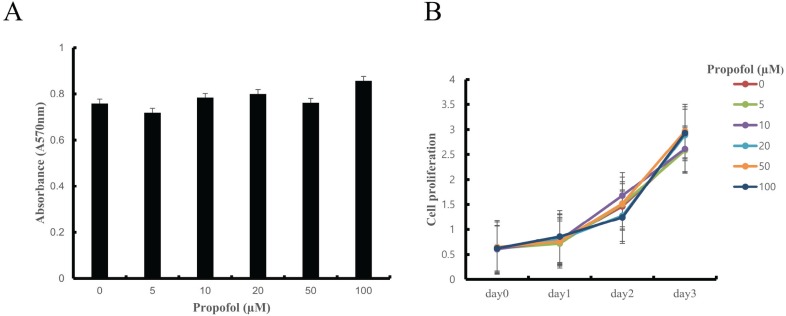
In previous studies, propofol concentrations of 30 and 100 µM have shown cytoprotective effects against stress-induced osteoblast cell death in culture [13]. Propofol at 30 µM mimics clinical plasma concentrations [14]. On this basis, we used propofol in the range of 0–100 µM. To examine the cytotoxicity of propofol on BMMs, cells were cultured in osteoclastogenic medium containing propofol (0–100 µM) for 3 days followed by MTT assay. Propofol did not have any cytotoxic effects on BMMs. Cell proliferation also showed no significant differences at the various propofol concentrations (Fig. 1A and 1B).
2. Propofol enhances osteoclastogenesis and osteoclastic bone resorption
To examine osteoclast formation, BMMs were cultured in an osteoclastogenic medium with various concentration of propofol for 4 days. Propofol increased the percentage of TRAP-positive multinucleated osteoclasts in a dose-dependent manner (Fig. 2A). Addition of propofol to the BMMs cultured with RANKL significantly increased the number and size of osteoclasts in a dose-dependent manner, with the maximum increase observed at 50 µM (Fig. 2B and 2C), implying that propofol enhances osteoclast differentiation and maturation. To further evaluate the influence of propofol in the bone resorption activity of osteoclasts, we performed the bone resorption assay. This showed that propofol treatment (50 µM) led to a significant difference in augmentation of resorption pits on dentin discs, which act as substrates for bone resorption, as compared to the untreated dentin discs (Fig. 2D and 2E). Hence, for the subsequent experiments, 50 µM propofol was used for osteoclastogenesis.
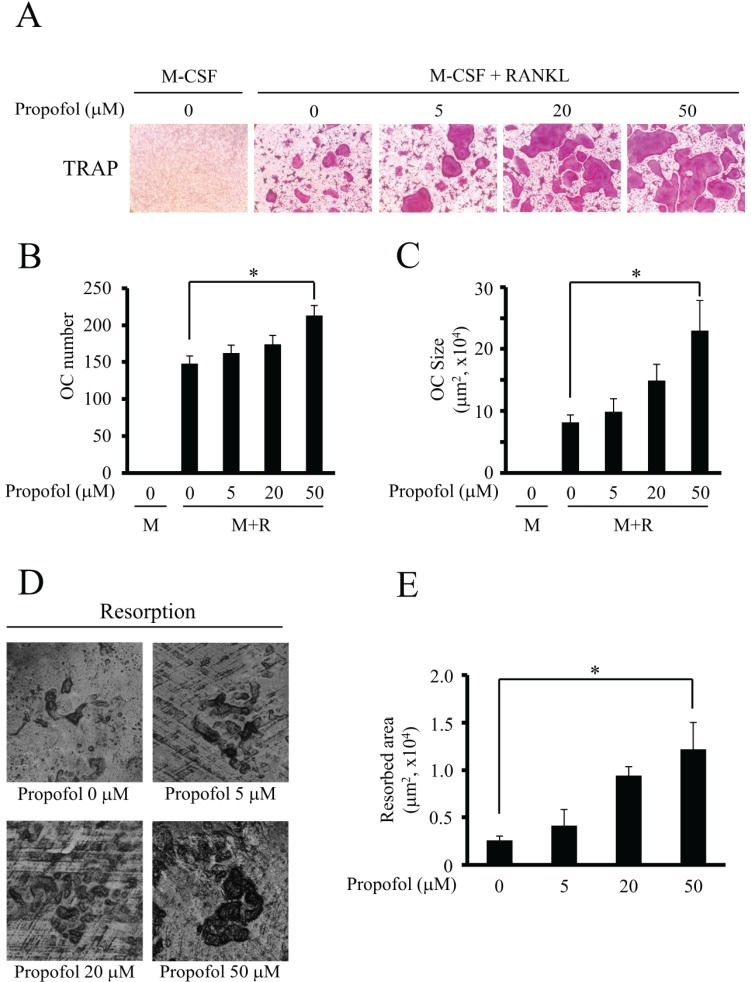 | Fig. 2Propofol markedly augmented the size of osteoclast and subsequently increased bone resorption. (A–C) BMMs were cultured with M-CSF alone or M-CSF plus RANKL with propofol (0–50 µM) for 4 days, and TRAP staining was performed on the resulting osteoclasts. (A) Example images are shown. (B) Quantification of TRAP-positive multinucleated osteoclasts (cells containing more than four nuclei). (C) Quantification of the size of osteoclasts. OC, osteoclast; M, M-CSF; M+R, M-CSF+RANKL. (D–E) To examine the resorption activities of osteoclasts, a bone resorption assay was performed using dentin discs. The resorption pits were visualized and quantified by laser scanning of cell-removed dentin discs. (*, P < 0.05).
|
3. Propofol does not affect the expression of osteoclastogenic transcription factors, c-Fos and NFATc1
The binding of RANKL to its receptor, RANK, triggers various signaling cascades, which culminate in the expression of the master transcription factor for osteoclastogenesis, NFATc1. NFATc1 is strongly induced by RANKL and its induction is necessary and sufficient for osteoclast differentiation. The induction of NFATc1 by RANKL is known to be regulated by intracellular Ca2+ oscillations and levels of another transcription factor, c-Fos [1516]. Since propofol significantly increased the formation of large osteoclasts, changes in the expression of NFATc1 and c-Fos were expected upon propofol treatment. As shown in Fig. 3A, the expression of c-Fos and NFATc1 was upregulated by RANKL (100 ng/ml) and the expression of c-Fos correlated with that of NFATc1. However, propofol (50 µM) did not affect the expression of c-Fos and NFATc1 at days 2 and 4. RT-PCR was performed to evaluate the mRNA expression levels of c-Fos, NFATc1, and TRAP at various doses of propofol (0–100 µM), with TRAP serving as an osteoclastic differentiation marker. The mRNA levels of c-Fos, NFATc1, and TRAP were not enhanced with increasing doses of propofol in the osteoclastogenic medium (Fig. 3B). In addition, western blotting indicated that the protein levels of c-Fos and NFATc1 did not intensify with increasing doses of propofol (Fig. 3C). Hence, we conclude that propofol-induced osteoclast formation is not mediated by changes in the expression of c-Fos and NFATc1.
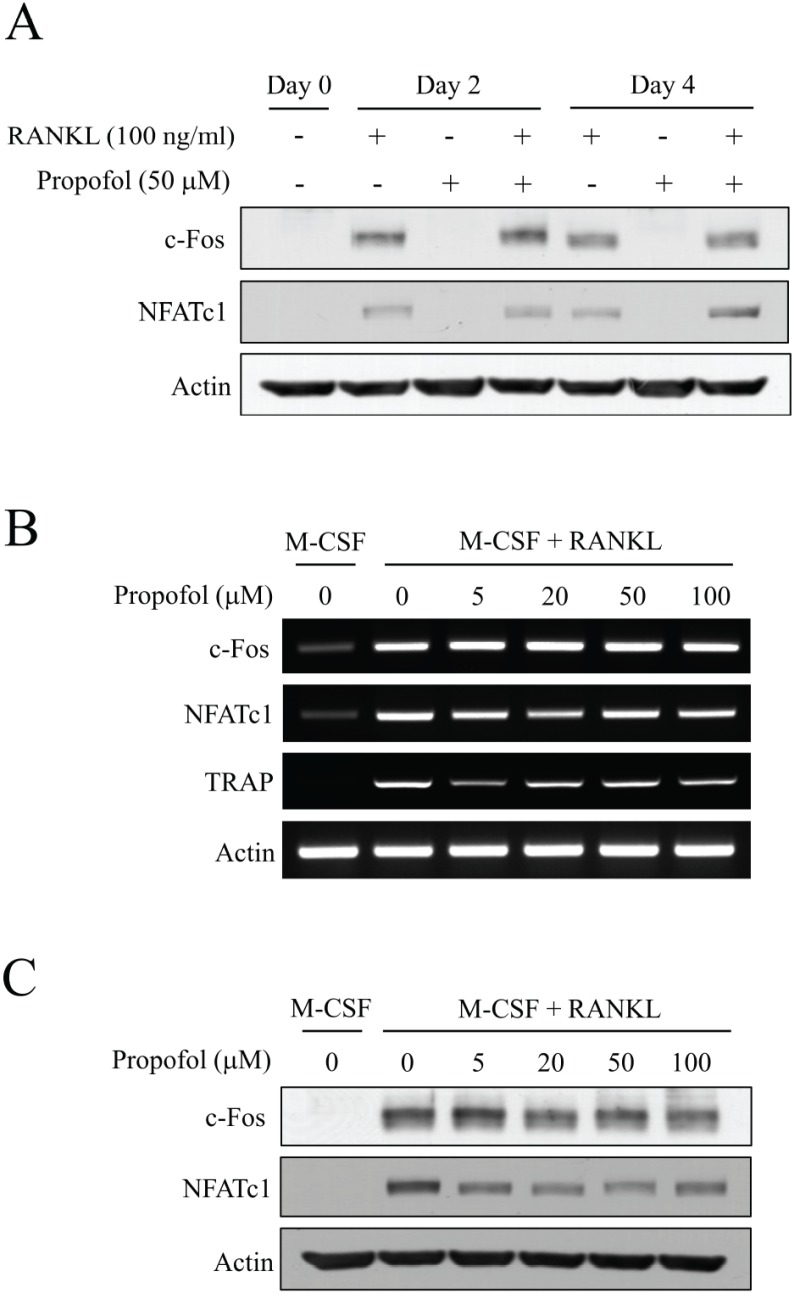 | Fig. 3Propofol has little effect on the expression of c-Fos and NFATc1. (A) BMMs were cultured in the presence or absence of RANKL (100 ng/ml) or propofol (50 mM) for 4 days. Expression of c-Fos and NFATc1 were evaluated by western blotting at day 0, 2 and 4. (B) The mRNA levels of c-Fos and NFATc1 were evaluated by RT-PCR. The mRNA expression of TRAP served as an osteoclastic differentiation marker. (C) The protein expression of c-Fos and NFATc1 at various concentrations of propofol (0–100 µM) was determined by western blotting.
|
4. Propofol increases osteoclast cell-cell fusion through p38 mediated DC-STAMP expression
The activation of the mitogen-activated protein kinase (MAPK) signaling pathway by RANKL is an important mechanism involved in osteoclastogenesis, and p38 kinases are an important subclass of MAPK signaling intermediates [1718]. To explore whether propofol-induced osteoclast differentiation is mediated by the activation of p38, the phosphorylation status of p38 in BMMs cultured in osteoclastogenic medium was assessed through western blotting. Propofol treatment (50 µM) caused p38 activation as shown by an increase in p38 phosphorylation (Fig. 4A).
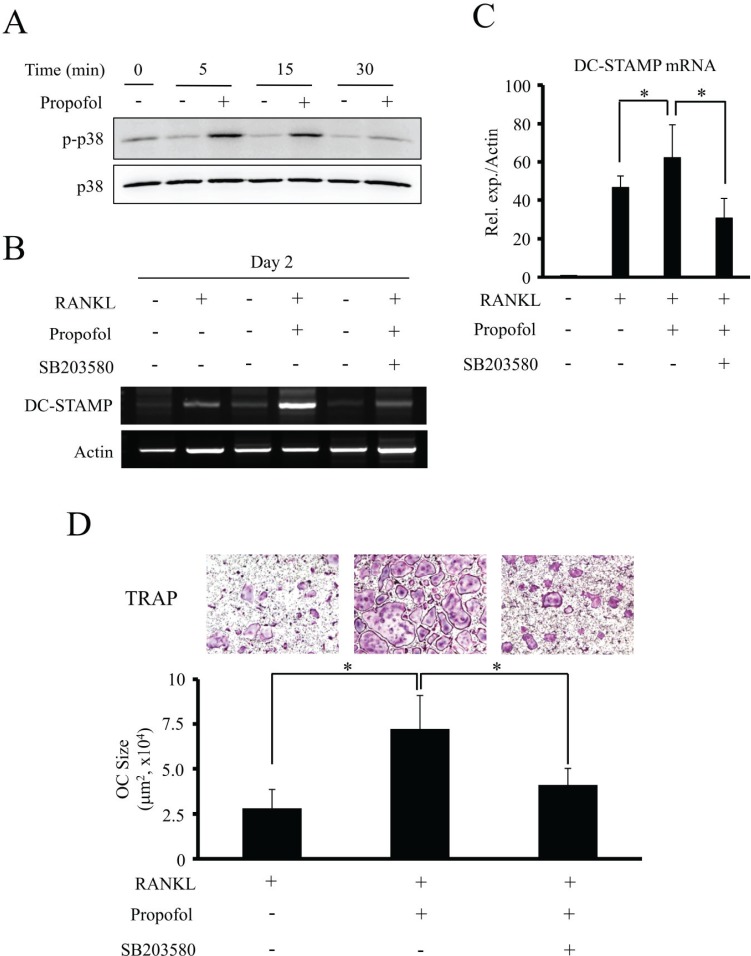 | Fig. 4Propofol increases osteoclast size through the activation of p38 and DC-STAMP expression. (A) Following incubation of BMM cells with propofol for the indicated times, phosphorylation of p38 was evaluated by western blotting. (B) The mRNA expression of DC-STAMP was analyzed by RT-PCR after treatment with the indicated combinations of RANKL (100 ng/ml), propofol (50 µM) and a p38 inhibitor, SB203580 (20 µM) for 2 days. (C) Quantitative real-time PCR analysis of DC-STAMP. (D) The representative TRAP-stained images are shown (top) and the sizes of mature osteoclasts were measured (bottom). Data are mean ± SD (*, P < 0.05). OC, osteoclast.
|
To investigate how propofol upregulates the production of multinucleated osteoclasts, we examined its effect on DC-STAMP, a protein essential for osteoclast cell-cell fusion. BMMs were cultured in the presence or absence of RANKL (100 ng/ml) and propofol (50 µM) for 2 days. In addition, we pretreated BMMs with the p38 inhibitor, SB203580 (20 µM) to verify whether the induction of DC-STAMP by propofol is mediated through p38 signaling. The DC-STAMP transcript levels, as analyzed by quantitative real-time PCR and RT-PCR, showed maximum upregulation upon a combined treatment of RANKL and propofol. However, SB203580 significantly reduced the propofol/RANKL-induced increase in mRNA levels of DC-STAMP (Fig. 4B and 4C). As shown in Fig. 4D, TRAP-positive multinucleated osteoclasts were increased by propofol treatment. A combined treatment of propofol with RANKL caused a significant increase in the size of mature osteoclasts as compared to RANKL alone. Formation of multinucleated osteoclasts and the size of mature osteoclasts showed a marked reduction upon p38 inhibition by SB203580, indicating that propofol treatment enhances osteoclast cell-cell fusion via DC-STAMP expression, mediated by p38 activation.
Go to :
DISCUSSION
Osteoclasts are involved not only in bone resorption but also in the cross-talk with osteoblasts and other cell types to regulate bone remodeling, which is essential for maintaining skeletal integrity [819]. We examined the effect of propofol on osteoclastogenesis and osteoclastic bone resorption, and the mechanism underlying this effect. In this study, propofol was found to enhance RANKL-induced osteoclast differentiation and maturation and, consequently, osteoclastic bone resorption, without affecting the expression of the osteoclastogenic transcription factors, c-Fos and NFATc1. Propofol also promoted osteoclast cell fusion by increasing DC-STAMP expression via p38-mediated signaling.
Understanding the regulatory mechanisms of osteoclastogenesis is a prerequisite for exploring the role of propofol on osteoclast differentiation and activation. The binding of RANKL to its receptor, RANK, leads to the recruitment of the TNF receptor-associated family proteins, which activate downstream signaling pathways mediated by MAPKs, JNK, ERK, and p38 [13]. These MAPKs induce the upregulation of c-Fos and NFATc1, which are the key transcription factors for osteoclast differentiation [20]. In particular, p38 is crucial to RANKL-induced osteoclast differentiation and activation, due to its involvement in the expression of several osteoclast-specific genes. Stimulation of p38 activates the microphthalmia-associated transcription factor (Mitf) and promotes the expression of TRAP and cathepsin K (CTSK) [212223]. These genes were also reported to be the downstream transcriptional targets of NFATc1 [24]. The activation of TRAP and CTSK leads to osteoclast differentiation and development of mature osteoclasts [2122]. Mature and active osteoclasts are generated by activation of various transcription factors such as NF-κB, c-Fos, PU.1, Mitf, and NFATc1 [2526]. In our study, propofol treatment increased the number and size of osteoclasts without altering the expression of c-Fos and NFATc1. NFATc1 has been implicated as the major regulator of osteoclast differentiation and the overexpression of active NFATc1 leads to osteoclast differentiation even if RANKL is absent [1627]. At the final stage of differentiation, NFATc1 Mitf, and PU.1 act synergistically to activate osteoclast target promoters [2829]. In this study, although the propofol-induced increase in osteoclast differentiation was not accompanied by increased expression of NFATc1, it is possible that propofol affected the activity of NFATc1 or its coordination with Mitf and PU.1.
During osteoclastogenesis, osteoclast size is increased due to cell-cell fusion-mediated multinucleation, and this preosteoclast fusion results in the formation of mature osteoclasts that activate the resorption of larger areas of bone tissue [30]. Therefore, the cell-cell fusion of osteoclasts is an essential process for osteoclast activation. DC-STAMP is a key molecule for cell-cell fusion during osteoclastogenesis and it is upregulated by the RANK-LNFATc1 pathway [31]. In this study, we demonstrated that propofol treatment significantly increased DC-STAMP transcript levels and extended the size of multinucleated osteoclasts, and both of these effects were inhibited by the p38 inhibitor, SB203580. However, NFATc1 expression remained unaffected upon propofol treatment, suggesting that propofol aids osteoclast cell-cell fusion via the induction of DC-STAMP expression through p38 rather than NFATc1. This in vitro study, therefore, successfully provides cell-based evidence on which to base more clinically-focused studies.
DC-STAMP-knockout mice have been reported to exhibit bone healing retardation compared to wild-type mice. In addition, fracture patients exhibit elevated circulating DC-STAMP+ cells during bone healing [32]. These results suggest that DC-STAMP can contribute to bone repair. Upregulation of DC-STAMP expression upon propofol treatment may contribute to bone healing in fracture sites as well as bone remodeling, however further investigation is required to confirm this.
In a recent study, α-tocopherol, which is structurally similar to propofol, stimulated osteoclast fusion by inducing the expression of DC-STAMP through activation of p38 and Mitf, and caused increased bone resorption [9]. However, many other factors such as CD9, CD44, macrophage fusion receptor, E-cadherin, and meltrin-α have reported roles in osteoclast cell fusion [33]. Therefore, further studies are required to investigate the regulatory role of propofol on osteoclast cell-cell fusion mediated by these factors.
In conclusion, we report that propofol treatment promotes osteoclast differentiation and maturation, and subsequently, bone resorption. Additionally, we show that propofol aids in osteoclast cell-cell fusion through p38-mediated DC-STAMP expression (Fig. 5).
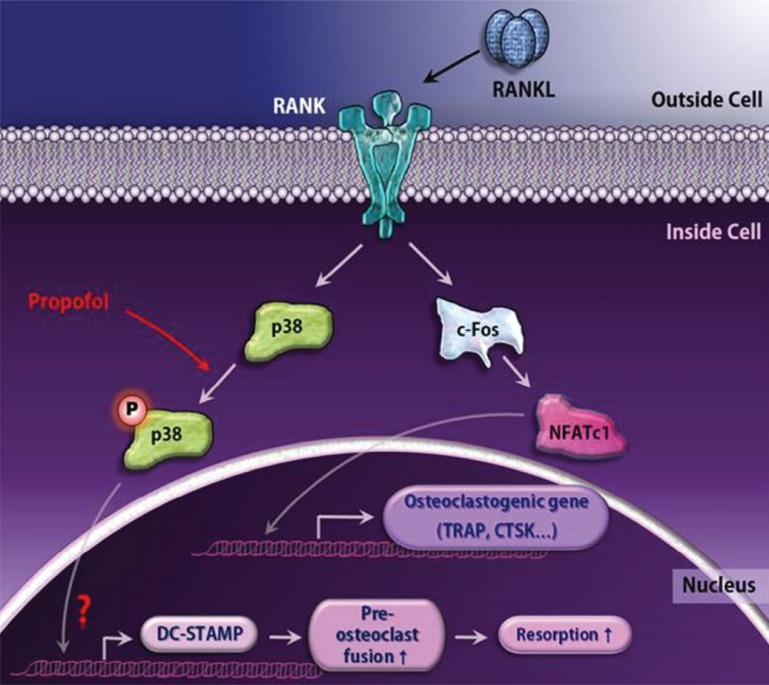 | Fig. 5A schematic diagram for propofol-induced large osteoclast formation. The engagement of RANKL to RANK leads to pivotal osteoclastogenic gene expression via the c-Fos/NFATc1 transcription factor axis. Although propofol does not alter these major differentiation pathways, it functions as a positive regulator of osteoclastogenesis by upregulating phosphorylation of p38 and subsequently increasing DC-STAMP expression.
|
Go to :
 XML Download
XML Download