

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
In patients with diabetes, hyperglycemia is induced by disrupted insulin secretion from pancreatic β cells or dysfunction in the metabolic action of insulin in various tissues [1]. Chronic hyperglycemia in patients with diabetes contributes to the development of macro- and microvascular complications [2]. Early intervention of hyperglycemia can inhibit or delay the development of various diabetes-related complications, such as nephropathy, retinopathy, and neuropathy [3]. The primary strategy to manage diabetes is modifying the diet pattern and physical activity of the patients. Numerous well-known oral anti-diabetic drugs, such as metformin, sulfonylureas, glinides, thiazolidinediones, and α-glucosidase inhibitors, have been used for the management and/or treatment of diabetes [4]. However, these oral drugs have several side effects, including weight gain, hypoglycemia, gastrointestinal intolerance, and fluid retention [5].
Sodium-glucose cotransporter 2 (SGLT2), the major glucose transporter responsible for reabsorbing approximately 90% glucose in the renal tissue, is expressed in the S1 segment of renal proximal tubules [67]. SGLT1 is the primary transporter for glucose and galactose absorption in the intestine [8]. SGLT1 is also expressed in the S3 segment of renal proximal tubule [9]. The remaining 10% of renal glucose not reabsorbed by SGLT2 can be reabsorbed by SGLT1 [10]. Recently, highly selective inhibitors of SGLT2 have been approved as oral anti-diabetic drugs that improve glycemic control by inhibiting the reabsorption of glucose filtered through the renal glomeruli [1112]. Although SGLT1 is a low-capacity glucose transporter [13], the activity of SGLT1 is elevated by a compensatory mechanism caused by the administration of SGLT2 inhibitors [14]. Similarly, in SGLT2-deficient mice, SGLT1 can reabsorb up to 70% of the filtered glucose under normal physiological conditions [15]. Empagliflozin, a selective SGLT2 inhibitor, inhibits glucose reabsorption by only 56% in wild-type mice, but reduces glucose reabsorption by 100% in SGLT1 and SGLT2 double-knockout mice [10]. Thus, the use of SGLT1/2 dual inhibitor designed to reduce glucose absorption in the gastrointestinal tract via SGLT1 inhibition and to reduce renal glucose reabsorption via SGLT1 and 2 inhibition has been increasingly gaining interest [1617]. Furthermore, the insulin-independent mechanism of SGLT2 or SGLT1 can induce an increase in urinary glucose excretion with minimal risk of hypoglycemia and body weight gain [1618].
HM41322 is a newly developed oral SGLT1/2 dual inhibitor. In this study, the pharmacological profiles of HM41322 were evaluated through in vitro and in vivo pharmacokinetic studies, and its glycemic control and urinary glucose excretory effects were compared to those of dapagliflozin, another marketed SGLT2 inhibitor, via multiple animal experiments.
METHODS
Chemicals
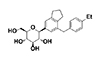
HM41322, {C24H30O5, (2S,3R,4R,5S,6R)-2-[7-(4-ethylbenzyl)-2,3-dihydro-1H-inden-5-yl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol} (Fig. 1) was synthesized by Hanmi Pharmaceutical (Hwaseong, Korea). Dapagliflozin, (1S)-1,5-anhydro-1-C-{4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl}-D-glucitol, was also synthesized by Hanmi Pharmaceutical (Hwaseong, Korea) according to a procedure in a previously published patent (patent no. WO2003099836 A1).
In vitro functional assay
HEK293 cells were purchased from the Korean Cell Line Bank (Seoul, Korea). Human SGLT1 and human SGLT2 cDNA were purchased from OriGene (Rockville, MD, USA). Stable cell lines expressing human SGLT1 and SGLT2 were established by transfecting HEK293 cells with vectors (pcDNA3.1(+)-FLAG-hSGLT1 and pcDNA3.1(+)-FLAG-hSGLT2), respectively, and selecting the positive clones following G418 treatment. The materials were assayed for their ability to inhibit [14C]-AMG (14C-methyl-α-D-glucopyranoside) uptake. IC50 values were calculated using a curve-fitting program (GraphPad, La Jolla, CA, USA).
Ethics statement for animal experiments
All animal experiments were approved by the Hanmi Research Center Institutional Animal Care and Use Committee and were carried out in accordance with the “Guide for the Care and Use of Laboratory Animals” (National Academies Press, 1996) and our institutional standard operating procedures (SOPs). The approval numbers were AECN0148 and SOP-TE094 for pharmacokinetic and pharmacology studies, respectively. Mice aged 7–8 weeks were housed in polysulfone cages (W×D×H=375×470×210 mm). The temperature (22±2℃) and relative humidity (50±20%) of the rodent housing environment were automatically controlled and recorded every 30 min. The lighting intensity was maintained at 150–300 Lux with a 12-h light and dark cycle. The feed and water were provided ad libitum.
Pharmacokinetic profiles
The pharmacokinetic profiles of HM41322 and dapagliflozin were evaluated in male C57BL/6 mice (n=2–3 per time point). The mice were provided free access to water and feed. The mice were fasted overnight before drug administration. For the oral pharmacokinetic analysis, HM41322 (0.3, 1, and 3 mg/kg) and dapagliflozin (1 mg/kg) were administered via oral gavage. For the intravenous pharmacokinetic analysis, HM41322 (1 mg/kg) and dapagliflozin (1 mg/kg) were intravenously injected. The vehicle contained 5% 1-methyl-2-pyrrolidinone, 20% PEG400, and 20 mM sodium diphosphate. Time points of sample collection were 0, 0.5, 1, 2, 4, 7, and 24 h after oral administration and 0, 0.08, 0.25, 0.5, 1, 2, 4, 7, 24 h after intravenous administration. Plasma samples were analyzed using liquid chromatography-mass spectrometry with a Waters Synapt G2 UPLC/QTOF (Waters Corporation, Wilmslow, UK). Pharmacokinetic values were analyzed using the Phoenix WinNonlin software (version 1.0, Certara L.P., St. Louise, MO, USA).
Urinary glucose excretion in mice
The effect of HM41322 on urinary glucose excretion was determined in eight-week-old male C57BL/6 mice. Three mice were assigned to each group according to their body weight. After overnight fasting, the mice were orally administered a single dose of dapagliflozin (1 mg/kg) or HM41322 (0.3, 1, or 3 mg/kg) and 2 g/kg glucose. After administration, the mice were moved into metabolism cages (Techniplast, Italy) for urine collection over 24 h. Urine volumes were measured, and urine samples were analyzed using a Hitachi 7020 (Hitachi USA). The urinary glucose and volume were normalized per body weight. According to the FDA new drug application for dapagliflozin (NDA 202293), the increased level of SGLT2 was solely inhibited by dapagliflozin at 1 mg/kg in C57BL/6 wild-type mice. A similar level of urinary glucose excretion was observed in both WT and intrinsic SGLT2 knock-out mice treated with dapagliflozin at 1 mg/kg. Therefore, dapagliflozin at a dose of 1 mg/kg was selected for comparing the in vivo pharmacological profile of SGLT1 and 2 dual inhibitor in male C57BL/6 mice.
Oral glucose tolerance and total glucagon-like peptide-1 determination in plasma in mice
Male C57BL/6 (8 weeks old) were fasted for 16 h before glucose loading. Dapagliflozin (1 mg/kg) or HM41322 (0.3, 1, or 3 mg/kg) was orally administered to the mice 30 min before glucose loading (2 g/kg, 10 ml/kg). Blood glucose levels were determined using OneTouch® Ultra strips (Life Scan, Inc., USA) and tail-nick blood collected during, prior to, and 0, 15, 30, 60, and 120 min after glucose loading. Glucose AUC0–120min was calculated via the oral glucose tolerance test (OGTT) using the trapezoidal rule. Inhibiting SGLT1 improves glucose control by reducing intestinal glucose absorption and stimulating glucagon-like peptide-1 (GLP-1) release [19]. Thus, for determining total GLP-1, plasma was collected via the subclavian vein at 30 min after glucose loading. GLP-1 was analyzed using an ELISA kit for multispecies (Millipore, Cat no. EZGLP1T-36K).
Blood glucose, HbA1c, and glucose tolerance after a 4-week administration in db/db mice
Eight-week-old male db/db mice were purchased from Charles River Japan. Seven mice were assigned to each group according to their glycosylated hemoglobin (HbA1c) and blood glucose levels after 16 h of fasting. Dapagliflozin (1 mg/kg/day) or HM41322 (0.3, 1, or 3 mg/kg/day) was orally administered for 28 days. The general clinical signs were observed at all dosing periods. HbA1c was analyzed using the drop collected from each cut of the tail vein at days 0 and 28 using a DCA vantage analyzer (Siemens, GER). Blood glucose level was measured using the tail vein blood drop at days 0, 3, 7, 10, 14, 17, 21, 24, and 28. At day 29, the OGTT was proceeded. Blood samples were collected at −60, 0, 15, 30, 60, and 120 min, which are similar to the time points of the single OGTT following glucose loading (2 g/kg). Body weight was measured daily by using a BSA224S-CW balance (Sartorius, GER).
Statistical analysis
Data in all tables and figures are presented as the mean±standard error of the mean (S.E.M.). Group data were analyzed using GraphPad Prism v.6.0 software (GraphPad Software, Inc., La Jolla, CA, USA) by one-way analyses of variance followed by Dunnett's multiple comparison test or by unpaired Student's t-tests.
RESULTS
In vitro potency and selectivity of HM41322 for SGLT1 and SGLT2
The chemical structure of HM41322 is shown in Fig. 1. HM41322 inhibited glucose transport in HEK293 cells with stable overexpression of human SGLT1 or SGLT2. As shown in Table 1 and Fig. 2, HM41322 inhibited glucose transport mediated by SGLT1 and SGLT2. The IC50 values for these transporters were 54.6 nM and 5.6 nM, respectively. However, although dapagliflozin, a well-known SGLT2 inhibitor, showed a high selectivity for SGLT2 (IC50=2.9 nM), it was less selective against SGLT1 (IC50=920.4 nM).
Pharmacokinetics in normal mice
The pharmacokinetic characteristics of HM41322 in the mice are summarized in Table 2 and Fig. 3. In mice treated with HM41322 at 0.3, 1, and 3.0 mg/kg, the area under the curve (AUC0–24h) values were 472.8, 1298.0, and 5456.1 ng·h/ml, respectively. The maximum concentration (Cmax) values were 142.3, 439.3, and 1830.0 ng/ml, respectively. Time to maximum concentration (Tmax) values were 0.5, 0.5, and 0.5 h, and half-life (T1/2) values were 3.1, 3.0, and 3.1 h, respectively. The AUC0–24h, Cmax, Tmax, and T1/2 values of dapagliflozin at 1 mg/kg were 457 ng·h/ml, 207 ng/ml, 0.5 h, and 3.7 h, respectively. The oral bioavailabilities of HM41322 based on the AUC0–24h values were 89% in the 1.0 mg/kg group and exceeded 100% in the other groups. However, the oral bioavailability of dapagliflozin was 67%. HM41322 showed excellent oral bioavailability (>89%) at all doses. These findings indicate that HM41322 has a good pharmacokinetic profile, high oral exposure, and approximate linear pharmacokinetics in normal mice.
Urinary glucose excretion in normoglycemic mice
Table 2 shows the effects of HM41322 and dapagliflozin on urinary glucose excretion for 24 h in normoglycemic mice. HM41322 induced a dose-dependent increase in urinary glucose excretion over the 24 h after dosing. The urinary glucose excretion level by the single administration of HM41322 (3 mg/kg) was higher than that of dapagliflozin (1 mg/kg).
Oral glucose tolerance in normoglycemic mice
In the OGTT, the blood glucose levels reached 285 mg/dl at 15 min after glucose loading. However, in the following 60 min, they decreased to 164 mg/dl and continued to reach an average of 76 mg/dl at 120 min post glucose loading. However, HM41322 suppressed the elevation in blood glucose levels in a dose-dependent manner throughout the course of the study (Fig. 4A). In Fig. 4B, the area under the curve (AUC) of glucose tolerance of the HM41322-treated groups (HM41322 0.3 mg/kg, 303.3±60.17 h·mg/dl; HM41322 1 mg/kg, 228.5±16.96 h·mg/dl; and HM41322 3 mg/kg, 170.7±13.14 h·mg/dl) was significantly lower than that of the vehicle control group (330.8±32.08 h·mg/dl). Similarly, dapagliflozin also decreased blood glucose increases during OGTT.
Plasma GLP-1 levels in normoglycemic mice
As shown in Table 3, the total GLP-1 plasma level in the HM41322-treated groups (HM41322 0.3 mg/kg, 15.0±6.6 pM; HM41322 1 mg/kg, 18.0±5.0 pM; and HM41322 3 mg/kg, 34.2±9.7 pM) increased in a dose-dependent manner, and was higher than that of the vehicle control group (11.4±3.0 pM). Dapagliflozin did not significantly increase GLP-1 plasma levels (15.5±5.7 pM).
Blood glucose, HbA1c, and glucose tolerance after a 4-week treatment of HM41322 in db/db mice
The effect of long-term treatment with HM41322 and dapagliflozin on glycemic control was evaluated in a 4-week repeat-dose study using db/db mice. HM41322 and dapagliflozin did not induce a significant change in body weight compared to that observed in the vehicle-treated db/db mice (Fig. 5A). From baseline to 4 weeks, the mean blood glucose level was increased in the vehicle control group, whereas HM41322 suppressed the elevation in blood glucose level throughout the course of the study in a dose-dependent manner (Fig. 5B). HbA1c, a marker of long-term blood glucose level, was significantly decreased following a 4-week treatment with HM41322 at all tested doses (Table 4). After 4-week repeated administration, the effect of HM41322 on the elevated post-prandial blood glucose was determined via OGTT. Consistent with the above data, HM41322 prevented an increase in plasma glucose levels in a dose-dependent manner (Fig. 5C). The AUCs of all HM41322-treated groups were significantly lower than those of the vehicle control group (Table 4).
DISCUSSION
Several SGLT2 inhibitors, such as empagliflozin [20], canagliflozin [21], and dapagliflozin [22], have been clinically used as antidiabetic drugs. Numerous reports have provided evidence to support their usefulness in reducing hyperglycemia by increasing urinary glucose excretion, both in nonclinical animal models [1823] and in patients with type 2 diabetes [1124]. All these drugs have selective inhibitory activities for SGLT2. Despite a major contribution of SGLT2 to renal glucose reabsorption, the application of selective SGLT2 inhibitors increases urinal glucose excretion by only 50–70% [25]. Because SGLT1 can compensate for the lack of SGLT2, absolute urinary glucose excretion was observed in both SGLT1- and SGLT2-deficient mice [18]. In addition, inhibitors of SGLT1 were recently shown to decrease blood glucose excursion after an oral glucose challenge, suggesting that such inhibitors may also be effective antidiabetic agents [2627]. Therefore, HM41322 was developed as a novel oral agent that is a dual inhibitor of SGLT1 and SGLT2. In the present study, in vitro pharmacodynamics, pharmacokinetic, and pharmacologic profiles of HM41322 were compared to those of dapagliflozin, a well-known highly selective SGLT2 inhibitor. In the in vitro selectivity study, HM41322 exerted high selectivity for human SGLT1 and SGLT2, with a 10-fold higher selectivity for SGLT2 than for SGLT1. HM41322 had a similar inhibitory effect on SGLT2 as dapagliflozin but, unlike dapagliflozin, it also simultaneously inhibited SGLT1. These results indicate that HM41322 was a selective dual inhibitor of SGLT1 and SGLT2, suggesting that it will exert potent inhibitory effects against SGLT1 and SGLT2 in preclinical and clinical studies.
In the pharmacokinetic study in mice, the maximum circulating concentrations of HM41322 after single oral doses at 1 and 3 mg/kg were 439 and 1830 ng/ml, respectively, with a T1/2 of 3.1 h. Similar to that of dapagliflozin (Cmax; 457ng/ml), the blood level of HM41322 was sufficient to inhibit SGLT2 and SGLT1. HM41322 was rapidly absorbed and reached the circulation within 30 min (Tmax: 30 min). Indeed, HM41322 could rapidly control postprandial hyperglycemia in both normoglycemic and diabetic mice.
All inhibitors of SGLT2 were designed to increase urinary glucose excretion. Our study showed that oral administration of HM41322 maximized urinary glucose excretion by inhibiting both SGLT1 and SGLT2 in the kidney and might additionally delay glucose absorption in the intestine by inhibiting SGLT1. Indeed, the maximum urinary glucose excretion was 19.32±1.16 mg/g body weight at 24 h after a single dose of 3 mg/kg HM41322. Although this effect was observed in the highest dosage of HM41322, HM41322 may be more potent than dapagliflozin (10.70±1.16 mg/g body weight). Similarly, sotagliflozin, a well-known dual inhibitor of SGLT 1 and SGLT2, also showed greater improvements in blood glucose control and HbA1C levels than selective SGLT2 inhibitors in a preclinical study [28]. These results suggest that the relatively strong effect of HM41322 on urinary glucose excretion may be due to its dual inhibitory activities against SGLT1 and SGLT2 in the kidney.
In this study, total plasma GLP-1 level in HM41322-treated mice was also increased after oral glucose, but not in dapagliflozin-treated mice. Consistent with this finding, a recent study showed that an increase in postprandial plasma GLP-1 level was observed after meal challenge in SGLT1-/- mice but not in SGLT2-/- mice [19]. SGLT1 is the primary intestinal transporter for glucose absorption [8]. Intestinal glucose after meal rapidly increases plasma GLP-1 level through direct stimulation of L cells [29]. Inhibiting SGLT1 improves glucose control by reducing intestinal glucose absorption and stimulating GLP-1 release [19]. These finding suggested that HM41322 mediated an increase in plasma GLP-1 by inhibiting intestinal SGLT1, not intestinal SGLT2. Because GLP-1 increases insulin release from the pancreatic β-cells (the incretin effect) [30], HM41322 may have an additional benefit in glucose control.
On the basis of these in vitro and in vivo pharmacodynamic, pharmacokinetic, and pharmacological properties, the antidiabetic effect of HM41322 was further evaluated in normal and diabetic animal models. HM41322 significantly reduced glucose excursion during the OGTT in both normoglycemic and diabetic mice. In the long-term efficacy study using db/db mice, after 4 weeks of repeated oral dosing, HM41322 induced a significant reduction in HbA1c. In addition, HM41322 (3 mg/kg) was superior in reducing HbA1c than dapagliflozin (1 mg/kg). Collectively, our data showed that HM41322 improved glucose tolerance by inhibiting both SGLT1 and SGLT2. This mode of action of HM41322, unlike dapagliflozin, was consistent with previous studies in both SGLT1 and SGLT2-deficient mice [18] and another SGLT1 and SGLT2 dual inhibitor, sotagliflozin, in humans and animals [151628]. Although HM41322 has a beneficial effect on the blood glucose control and plasma GLP-1 release by blocking both SGLT1 and SGLT2, this benefit was observed only when HM41322 was administered at higher dosage (3 mg/kg) compared to dapagliflozin. Inhibiting SGLT1 by HM41322 can reduce intestinal glucose absorption and stimulates GLP-1 release. However, GLP-1 plasma levels were not significantly different between two groups of mice treated with HM41322 and dapagliflozin at the same dosage of 1 mg/kg. No difference between two agents on plasma GLP-1 levels can be explained by its selectivity for SGLT1 or SGLT2. Dapagliflozin has been known as a selective SGLT2 inhibitor. However, dapagliflozin also has a relatively weak inhibitory activity for SGLT1, and its selectivity for SGLT2 is weaker than other SGLT2 inhibitors such as empagliflozin and tofogliflozin, which are highly selective SGLT2 inhibitors [1431]. More interestingly, empagliflozin, a highly selective SGLT2 inhibitor, has also recently been shown to increase circulating GLP-1 [32], although the effect was relatively mild. The detailed mechanisms are not fully apparent, but the mechanisms underlying circulating GLP-1 elevation by the selective SGLT2 inhibitors are complex, and the multiple mechanisms, including those independent of SGLT1 inhibition, may be involved. Our data suggest that a higher dosage of HM41322 is needed to achieve the additional effect of dual inhibition of SGLT1 and SGLT2 using HM41322.
In summary, HM41322 was a potent dual inhibitor of SGLT1 and SGLT2 and, unlike dapagliflozin, which inhibited only SGLT2, it showed additional benefits on urinary glucose excretion and glucose intolerance in various animal models through simultaneous inhibition of SGLT1 and SGLT2. In the view of postprandial glucose control, dual inhibitors of SGLT1 and SGLT2 are more advantageous than inhibitors of SGLT2 alone because the former inhibits the absorption of intestinal glucose [8] and also affects GLP-1 secretion. On the basis of these findings, additional SGLT1 inhibition might have benefits that could not be achieved with SGLT2 inhibition alone. Thus, the preclinical characteristics and pharmacokinetics of HM41322 supported its progression into clinical trials and served as an evidence of the additional benefit of dual inhibition of SGLT1 and SGLT2.








 XML Download
XML Download