

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Ischemia/reperfusion injury (IRI) caused myocardial detrimental damage by following blood restoration after coronary occlusion [1]. The most crucial inducers were calcium overloading and excessive ROS produced by mitochondria whose oxidative phosphorylation lost its normal function after reperfusion [2]. One of the clinical/experimental interventions was ischemic preconditioning (IPC) which targeted to mitochondrial ATP-sensitive potassium channel (KATP channel), having been found could reduce the infarct size of heart left ventricular exposed to (IRI) [3]. Mechanism study had uncovered that selectively activating KATP channel by pharmacological intervention displayed the approximately effects of IPC in alleviating IRI [4]. Although IPC decreased ROS production after IRI, heart was an energy-consumption organ that demanded other energy metabolism for sufficient power supplying.
Ketone body was considered as a significant contributor to cellular and organic energy metabolism in biological states. Especially in pathological condition, ketone body acted as an important energetic source in mammalians when oxidative phosphorylation was damaged. Ketone body, consisted with 3-hydroxybutyrate (BHB), acetoacetate (ACAC) and acetone, were produced in liver and diffused to bloodstream [5]. Insight into cardiomyopathy, reduction of ketone body oxidation had been found and promoted pathological progress while injection of BHB before ischemia displayed a cardioprotective effect which conferred to reduce the infarct size and cell death [56]. In aspect of I/R injury, an increased uptake of ketone body was found in cerebral as stress-induced energy substrate, but just sustained for several minutes [7]. That indicated in the early on-set of I/R injury, short-term upregulation of ketone body metabolism served as energy supplementary. However, it was not enough for maintaining in the long-last period of I/R injury. Whether extra-providing ketone body enabled to preserve cardiac function when experienced I/R injury or other cardiomyopathies.
Studies of Parkinson's diseases showed BHB exhibited neuroprotective effects via mitochondrial respiratory and ATP production improvement [8]. Moreover, BHB was served as an endogenous and specific inhibitor of histone deacetylases that increased Foxo3a and Mt2 promotor histone acetylation, contributing to protection against oxidative stress [9]. In aspect of heart failure, BHB acted as not only an energy carrier but also a signaling regulator in cardiac pathological progress. Treatment of BHB apparently attenuated ROS production and cellular apoptosis in response to H2O2-induced oxidative stress [10]. ACAC, 3 mM, has also been served as a neuroprotective agent to retinal ganglion cells (RGC) against neurodegeneration induced by NMDA [11] and protects hippocampal neurons against glutamate-induced toxicity by energy metabolism-related machenisms [12]. In addition, SCOT-Heart-KO mice displayed more susceptible to myocardial pressure and oxidative stress, suggesting an essential role of SCOT-mediated ketone body signaling in cellular energy metabolism [13].
Nicorandil, a KATP channel opener, had been demonstrated exhibiting good cardioprotective effects, including reducing infarct size, improving the recovery of myocardial contractile function and preventing oxidative stress-induced cellular apoptosis [1415]. In addition, previous study had shown nicorandil attenuated IRI through PI3K/Akt signaling pathway to inhibit ER-stress-induced apoptosis [16]. However, whether nicorandil displayed the cardioprotective effects through the ketone body metabolism in H/R-induced cardiomyocytes was rarely studied.
In our study, we performed in vitro models to evaluate the effects of nicorandil on H/R-induced cytotoxicity. Nicorandil increased cellular and mitochondrial viability in H9c2 cells exposed to H/R. Meantime, nicorandil decreased ROS production, alleviated calcium overloading and reduced cellular apoptosis in mitochondria-mediated proapoptotic pathway. In crucial, nicorandil pretreatment upregulated protein and gene expressions of ACAT1 and OXCT1, and either of ketone body metabolism, which promisingly characterized the potential activities of KATP channel openers on ketone body metabolism.
METHODS
Cell culture
Rat H9c2 cardiomyocyte cell line was purchased from the American Type Culture Collection (CRL1446, ATCC, USA). H9c2 cells were cultured in high glucose (4,500 mg/l) Dulbecco's Modified Eagle Medium (DMEM) composed of 10% fetal bovine serum (FBS) and 1% v/v 100 U/ml penicillin and streptomycin (Gibco, Oklahoma, USA) in a humidified incubator containing 5% CO2 at 37℃.
Pharmacological interventions
For cellular treatment, nicorandil (Sigma-Aldrich, St. Louis, MO, USA) pretreated for 24 h and H/R were performed when cells reached 80–90% confluency. H9c2 cells were divided into 5 groups: i) control, cultured in normoxia (20% O2, 5% CO2); ii) H/R alone; iii) H/R+nicorandil (25 µM); iv) H/R+nicorandil (50 µM); and v) H/R+nicorandil (100 µM).
In vitro model of H/R
Hypoxia in H9c2 cells was induced by replacing the normoxia medium with Krebs-Ringer Bicarbonate (KRB) buffer that consisted of 115 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 24 mM NaHCO3, 1.2 mM KH2PO4, 1.2 mM MgSO4, 10 mM Hepes and 0.01% BSA, and placing cells into a chemical hypoxia system (BD, USA) that consumed oxygen rapidly to induce hypoxic condition. After 4 h hypoxia, H9c2 cells were subjected to reoxygenation by replacing the KRB buffer with cultural medium for 12 h.
Cell viability
Cell viability of H9c2 cells was determined by measuring the absorbance of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphe-nyltetrazolium bromide (MTT) purchased from Sigma-Aldrich (St. Louis, MO, USA). Cells were seeded in 96-well microplates at a density of 1×104 cells per well. After pharmacological treatment, cells were exposed to H/R and 100 µl cultural medium containing MTT (5 mg/ml, Invitrogen, USA) at a final concentration of 500 µg/ml in PBS was added to the plates (10 µl/well) and incubated for an additional 4 h at 37℃. Then MTT solution was discarded and DMSO (150 µl) was added to each well to solu-bilize formazan crystals formed in viable cells. The absorbance was measure optical density at 570/650 nm on a Multi-Mode Detection Platform (SpectraMax Paradigm, Molecular Devices, USA). Cell viability of control group was considered as 100%.
Mitochondrial viability
H9c2 cells were seeded in the dark 96-well microplate at a density of 1×104 cells per well. Cells were treated as described above. After treatments, the mitochondrial viability was evaluated by the mitochondrial viability assay kit (ab129732, Abcam, USA). The fluorescence was detected at 590 nm with an excitation wavelength of 550 nm. Mitochondrial viability of control group was considered as 100%.
Apoptosis measurement
H9c2 were seeded in 6-well plates (2×106 cells per well). Cells in the different treatment groups were harvested and resuspended in PBS. Following centrifugation at 1,000×g for 5 min, detached cells were collected and incubated with Annexin V-Fluorescein isothiocyanate (FITC) coupled with Annexin V protein and propidium iodide (PI) (Sigma-Aldrich, St. Louis, MO, USA) at room temperature in the dark for 15 min. Immediately after gently pipetting, samples were analyzed by flow cytometry by performing with a 488-nm laser coupled to a cell sorter. Apoptotic cells were characterized by high Annexin V binding and high PI staining.
ROS and calcium measurement
Mitochondrial ROS was measured by the DCFDA Cellular ROS Detection Assay Kit (Invitrogen, USA) according to the commercial protocol. Cellular calcium overloading was measured by Fluo-4, AM (Invitrogen, USA) fluorescent probe staining according to commercial protocol. After H/R, cells were washed with PBS 1 time and digested by Trypsin-EDTA. Resuspended cells with cultural medium and incubated with DCFDA or Fluo-4 probe for 30 min at 37℃ in dark. The ROS production and calcium concentration in each group were detected using BD FACSAria III flow cytometer (BD, USA) by intensity of green fluorescence (Ex 488 nm/Em 535 nm).
Western blot
H9c2 cells were lysed in ice-cold RIPA buffer (20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerophosphate, 1 mM Na3VO4, 1 µg/ml leupeptin, Cell Signaling Technology, USA) with and proteinase inhibitor cocktail (Roche, Basel, Switzerland). The cell lysates were immediately centrifuged at 13,000 g for 15 min at 4℃ and the supernatants were boiled with loading buffer for 10 min. The concentrations of total proteins were quantified by the Bio-Rad protein assay kit (Bradford method) (Bio-Rad, CA, USA). Equal amounts of proteins (30 µg/lane) were separated by 10% SDS-PAGE gels and transferred to NC membranes (pore size: 0.45 µm, Pall, BioTrace). The membranes were blocked in 5% non-fat milk in TBST (Tris-buffer saline with 0.05% Tween 20) for 1 h and incubated with ACAT1 (1:1000, Abcam, UK), OXCT1 (1:1000, Santa Cruz, USA), Bax and Bcl-2 (1:1000, Cell Signaling Technology, USA) primary antibodies overnight at 4℃. β-Actin (Santa Cruz, USA) was used as the loading control. This process was followed by 3 times TBST washes for 15 min and incubation with the secondary fluorescent antibodies (1:5000, Odyssey, USA) for 2 h at room temperature in dark. The fluorescent signals of bands were detected by the Odyssey infrared imaging system (Belfast, USA). The densities of bands were normalized to Actin.
RNA extraction, reverse transcription, and quantitative real-time PCR
Total RNA was obtained from the cultured H9c2 cells using FavorPrepTM Total RNA purification mini kit (Favorgen Biotech Corp., Pingtung, Taiwan). Tissues were lysed and homogenized in 300 µl of lysis buffer (RLT Buffer; Qiagen, Basel, Switzerland), using the Fast Prep 24 lyzer (MPbio, Lucerne, Switzerland). Total RNA of tissue was isolated on spin columns with silica-based membranes (RNeasy Mini Kit; Qiagen), following the manufacturer's instructions. RNA was eluted with 30 µl of H2O. A small amount of purified RNA (1 µg) from cells was reverse transcribed in a volume of 20 µl using the RT High-Capacity RNA-to-cDNA Kit (Applied Biosystems, Rotkreuz, Switzerland). Synthesized cDNA was then stored at −20℃. Quantitative real-time PCR were performed with the ViiA™ 7 real-time PCR system (Applied Biosytems, Grand Island, NY, USA) using the Taq polymerase master mix FastStart Universal SYBR-Green Master Rox (Roche Diagnostics, Indianapolis, IN, USA) according to the manufacturer's instructions. Primer sequences quantified for mRNA were directed against ACAT1, OXCT1, as well as β-actin mRNA used as an endogenous control (primer sequences are listed in Table 1). The PCR mixture is comprised of 10 µl SYBR master mix, 0.5 µl forward primers, 0.5 µl reverse primers, 1 µg template and ddH2O makes up to 20 µl. The procedure of RCR is 50℃ for 2 min, 95℃ for 10 min, followed by 45 cycles of 95℃ for 15 s, 60℃ for 60 s. Gene expression level was based on the comparative CT value using the 2−ΔΔCT method and normalized with β-actin.
Cellular BHB and ACAC determination
Cellular BHB concentrations were determined using a Colorimetric Assay Kit (Cayman Chemical Company, USA). The Cells (~18×106 cells) were collected with a rubber policeman and then centrifugated at 2,000 g for 10 min, 4℃. Cell pellets were resuspended in 1–2 ml of cold assay buffer, then sonicated with 20× at 1 s bursts and centrifugated at 10,000 g for 10 min, 4℃. Supernatants were removed, the pellets were resuspended in 1 ml of cold assay buffer and stored on ice. Each sample was measured in triplicate with 50 µl of the sample in each well, and reacted by adding 50 µl of the developer solution. Then incubated the plate at 25℃ in the dark for 30 min and then read the absorbance at 445–455 nm using a microplate reader.
Cellular ACAC concentrations were determined using a Colorimetric Assay Kit (Abcam, UK). After different treatments, cell pellets were collected and resuspended with ddH2O. Sample volumes were adjusted to 110 µl, 80 µl ACAC assay buffer and 10 µl ACAC substrate were added and mixed well. After incubation at 25℃ for 15 min, the values of absorbance at OD 550 nm were measured in a kinetic mode.
Cellular succinate and acetyl-CoA determination
Cellular succinate concentrations were determined using a Colorimetric Assay Kit (BioVision, USA). In brief, H9c2 cells were homogenized rapidly and centrifuge at 10,000×g for 5 min. Collected the supernatants and mixed with reagents into 96-well plate. After 30 min incubation at 37℃, samples were measured at 450 nm. The values of control group were considered as 1.0.
Cellular ACAC concentrations were determined using a Colorimetric Assay Kit (Abnova, TaiWan) according to manufactory's protocol. H9c2 cells were homogenized rapidly on ice and centrifuge at 10,000×g. Supernatants were collected and neutralized pH to 6–8 and diluted volume to 50 µl. Then mixed samples with 50 µl reaction mix to be tested with fluorescent detection by using Ex/Em=535/589. The values of control group were considered as 1.0.
RESULTS
Nicorandil protected cardiomyocytes against H/R-induced cytotoxicity
To examine whether nicorandil had anti-H/R effect, we used a well-established H/R model that consisted of 4 h hypoxia followed by 12 h reoxygenation as shown in Fig. 1A. At first, MTT assays and mitochondrial viability assay were performed to evaluate cytotoxicity of nicorandil, and the control was used as a maximum reference (100%) to calculate the effects of nicorandil (25, 50, 100 µM) on cell and mitochondrial viability. After 24 h treatment, increasing concentrations of nicorandil (up to 100 µM) did not cause cellular cytotoxicity in H9c2 cells as shown in Fig. 1B. Consistently, different concentrations of nicorandil showed no significant effect on mitochondrial viability (Fig. 1C).
Subsequently, H/R significantly decreased the cell and mitochondrial viability over 50% compared with control group (100%), respectively. Increasing concentrations of nicorandil treatment enhanced cardiomyocytes survival after H/R in a dose-dependent manner, showing a maximum of 77% cell viability and 78% mitochondrial viability with 100 µM nicorandil (Figs. 1D and E).
Nicorandil suppressed cellular apoptosis and oxidative stress in H9c2 cells exposed to H/R
Quantitative analysis using flow cytometry further confirmed that the apoptotic index (percentage of cells in early apoptosis plus late apoptosis) was markedly increased in H/R group (more than 40%) compared with that of control group (less than 10%). However, a decrease in the apoptotic rate was observed in H9c2 cells pretreated with nicorandil (Figs. 2A and B). The effect of nicorandil at the concentration of 100 µM decreased apoptotic rate to less than 20%.
Next, mitochondrial ROS was measured by the DCFDA fluorescent probes staining. H9c2 cells exposed to H/R resulted in significantly enhancement of ROS level compared with the control group. Treatment of nicorandil at the concentration of 25 µM significantly suppressed ROS level, and nicorandil at the concentration of 100 µM abolished H/R-induced ROS generation in large extent (Fig. 2C). Moreover, investigation was employed to determine whether the cardioprotective effect of nicorandil was related to the suppression of intracellular calcium overloading after H/R. Fig. 2D showed a significant decrease of calcium concentration in nicorandil groups compared with H/R group.
To further explore the potential apoptotic signaling in protective effect of nicorandil against H/R-induced cytotoxicity, the expressions of Bax and Bcl-2 were measured by western blotting. As shown in Fig. 2E, compared with the normal group, the expression of Bcl-2 decreased in the H/R group while the expression of Bax did not change, showing the decreased ratio of Bcl-2 to Bax in H/R group. Pretreatment of nicorandil increased the protein expressions of Bcl-2 and the ratio of Bcl-2 to Bax dose-dependently, comparing with H/R group (Figs. 2E and F).
Nicorandil up-regulated ACAT1 and OXCT1 expressions in H9c2 cells exposed to H/R
To demonstrate the potential of nicorandil on ketone body metabolism, we evaluated the expressions of ACAT1 and OXCT1 in H9c2 cells induced by H/R. Firstly, immunoblot analysis showed decreasing protein levels of ACAT1 and OXCT1 in H9c2 cells induced by H/R. Interestingly, nicorandil significantly increased the protein levels of ACAT1 and OXCT1 in dose-dependent manner (Figs. 3A and B). Then we further confirmed with the mRNA expression of ACAT1 and OXCT1. After H/R, ACAT1 and OXCT1 mRNA expressions were significantly decreased. Changes in these mRNA levels were consistent with the up-regulation of protein levels, nicorandil enhanced mRNA expressions of ACAT1 and OXCT1 with a great extent comparing with H/R group (Fig. 3C).
Nicorandil up-regulated ketone body metabolism in H9c2 cells exposed to H/R
In addition, we performed detection of BHB and ACAC concentration to examine the effects of nicorandil in H/R-induced H9c2 cells. BHB concentration was decreased significantly in H/R group, and nicorandil up-reuglated BHB in H/R-induced H9c2 cells (Fig. 4A). Meanwhile, ACAT level reduced significantly in H/R group, followed by a significant augmentation in nicorandil group in dose-dependent manner (Fig. 4B). The combined analysis (ACAC+BHB) presented the similar decent when compared with the expressions of ACAT1 and OXCT1 that demonstrated a significant elevation of ketone body metabolism in nicorandil pretreated H9c2 cells in expose to H/R.
Nicorandil up-regulated succinate and acetyl-CoA production in H9c2 cells exposed to H/R
To further evaluate the activities of ACAT1 and SCOT1, we detected the concentrations of their metabolic products, succinate and acetyl-CoA. In cells exposed to H/R, the amounts of succinate and acetyl-CoA decreased significantly, while pretreatment of nicorandil significantly increased the amounts of succinate and acetyl-CoA, suggesting the upregulated expressions of ACAT1 and SCOT1 contributed to enhanced ketone body metabolism (Figs. 5A and B).
DISCUSSION
Nicorandil had been demonstrated protected heart and brain against ischemia/reperfusion injury by reducing cell apoptosis. As nicorandil is a mitochondrial KATP channel opener, it presents preconditioning effects and maintains energy metabolism when experiencing lethal injury. However, even if nicorandil could help mitochondria for their oxidative phosphorylation process, it was not enough for normal energy consumption, especially in reperfusion period, in which large amount of ROS produced and electron transport chain would be ineffective. Taking it for consideration, the reason why nicorandil can still display a good effect on I/R injury probably because of a supplementary energy metabolism system----ketone body metabolism. In our study, we used in vitro model of hypoxia/reoxygenation (H/R) in H9c2 cell line to mimetic in vivo model of ischemia/reperfusion injury. After experiencing 4 h hypoxia and 24 h reoxygenation, H9c2 cell viability decreased more than 50%, as well as mitochondrial viability, demonstrating it was applicable for the subsequent study. Pretreatment of nicorandil displayed cardioprotective effects evaluated by MTT assay and mitochondrial viability assay. To further evaluated whether nicorandil could reduce cellular apoptosis, we performed Annexin V/PI double staining assay to detected cellular apoptotic rate after H/R. Results analyzed by flow cytometry showed there was a significantly increasing population of apoptotic cells (over 40%), comparing with control group (less than 10%). Pretreatment of nicorandil decreased cellular apoptotic rate dose-dependent (less than 20% in 100 µM nicorandil treatment group). Taken together, our in vitro model of H/R major caused H9c2 cells death by inducing cellular apoptosis and nicorandil reduced myocardial injury by increasing mitochondrial viability and preventing cellular apoptosis.
Oxidative stress and calcium overloading were considered as two main hypotheses to explain the pathogenetic progress of ischemia/reperfusion injury. In general, oxidative stress, associated with extra-formation of reactive oxygen species (ROS), contributed to modification of phospholipids and proteins and further changed membrane permeability. That influences membrane ion channels, like Na+/K+ pump and Ca2+ pump, which impaired the balance of internal and external concentration of Ca2+, leading to calcium overloading [17]. Our in vitro model of H/R significantly increased cellular ROS production and caused calcium overloading in H9c2 cells. Pretreatment of nicorandil had the ability to reduce the excessive production of ROS and calcium overloading dose-dependently, indicating nicorandil protected cardiomyocytes against H/R by alleviating oxidative stress.
Beyond the theory that oxidative phosphorylation would lose its function for a long-term period even when oxygen was resupplied to cardiomyocytes [1], there was, doubtlessly, other energy production pathways to maintain cellular metabolism for cell survival. Whether improving these metabolisms' efficiency could address the issue of energy supplying to keep cell survival when experiencing H/R. One of energetic sources that feed cells in oxygen-existing condition is ketone body metabolism. Ketone bodies, served as supplementary energy source for energy-consumption tissues in times of fasting and exercise [23], were produced by fatty acids via mitochondrial β-oxidation in liver. The fatty acids were firstly metabolized to acetyl-CoA and then produced to ACAC, which was the precursor of the other ketone bodies, acetone and BHB. When taken up by heart, BHB will converted back into ACAC. Meantime, ACAC obtained a CoA donated by Succinyl-CoA to form acetoacetyl-CoA, catalyzed by OXCT1 (also known as SCOT). Then acetoacetyl-CoA were going to be converted to two acetyl-CoA by ACAT1 and participated into tricarboxylic acid cycle for ATP production [18]. Take insight into cardiac pathogenesis, while the role of fatty acids had been explored decades, the dynamics of ketone body metabolism in regulation of myocardial energy utilization and mitochondrial biogenetics were rarely studied when facing external stresses. Rebecca C. Schugar's study demonstrated ketone body oxidation play an important role in free radical homeostasis that promoted myocardial response to pressure overloading [13]. In the failing heart, energy demand was considered as the most important problem as well as the cardiac capacity to utilize other fuels, such as ketone body. In Gregory Aubert's study, they used quantitative mitochondrial proteomics to identify dysfunctional proteins during the development of heart failing. They found proteins involved in ketone body metabolism were downregulated in failing heart. Moreover, they uncovered that ketone bodies oxidation reduced heart failing in an ex vivo model of isolated perfused heart, indicating they acted as a crucial fuel source for energy metabolism [19]. In extent of failing human heart, ketone body utilization performed as an alternative fuel and adaptive metabolic products [20]. In our study, the protein expressions of ACAT1 and OXCT1 decreased in H9c2 cells exposed to H/R. Pretreatment of nicorandil significantly upregulated the protein expressions of ACAT1 and OXCT1 and either of their mRNA expressions. Further quantification of ketone bodies showed nicorandil also upregulated two major metabolic products, BHB and ACAC. Moreover, pretreatment of nicorandil significantly increased the amounts of succinate and acetyl-CoA, suggesting the upregulated expressions of ACAT1 and SCOT1 contributed to enhanced ketone body metabolism.
Beyond basic acknowledgement between ketone body and energy metabolism, novel insights of ketone body functions were discovered. Recent study showed gut microbiota increased ketone utilization during fasting by using functional genomic, biochemical, and physiologic assays, implying the benefit provided by gut microbiota in myocardial ketone body metabolism [21]. Ketone body also suppressed autophagic elimination in neurons to prevent cell death induced by glucose deprivation. In the presence of BHB, the level of LC3-II and p62 declined in neurons in expose to glucose deprivation [22]. Our finding gives a new insight into the effect of nicorandil on cardiac injury and cellular apoptosis via regulation of ketone body metabolism, by which these results enrich our view to better understand the complex energy metabolisms in heart when experiencing I/R injury and new strategy for drug discovery.



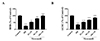
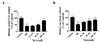

 XML Download
XML Download