

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Neuropathic pain is a chronic pain state that is caused by the dysfunction of the somatosensory nervous system. It is characterized by a series of neurobiological events such as unpleasant abnormal sensation (dysesthesia); an increased response to painful stimuli (hyperalgesia); and pain in response to stimuli that do not normally cause pain i.e., allodynia [123]. Peripheral neuropathic pain may result from damage to the peripheral nerves (sensory, motor or autonomic), which is generally observed in the patients with cancer, AIDS, long standing diabetes, lumbar disc syndrome, herpes infection, traumatic spinal cord injury, multiple sclerosis, stroke, thoractomy, herniorrhaphy, mastectomy and sternotomy [4]. Central neuropathic pain is a consequence of injury of the central nervous system (brain and spinal cord) that may result from spinal cord injury, stroke and multiple sclerosis [5]. Abnormal neuronal activity may develop at the site of injury, in dorsal horn of cell of the spinal cord or at various synaptic regions in the brain that participate in the processing of somatosensory information.
Opioids show limited clinically efficacy in neuropathic pain. Moreover, development of abuse potential and tolerance to pain also limit the common use of opioids in neuropathic pain. Tricyclic anti-depressants (TCAs) such as nortriptyline, desipramine (secondary amines) and amitriptyline, imipramine (tertiary amines) have been used in chronic neuropathic pain [6]. However, these have been largely replaced due to common side effects such as sedation, anti-cholinergic effects and orthostatic hypotension. Selective serotonin and norepinephrine reuptake inhibitors (SSNRIs) such as duloxetine and venlafaxine have also been used to provide pain relief [7]. Other antidepressants such as SSRIs (citalopram and paroxetine) have also shown efficacy in painful diabetic peripheral neuropathy [8]. However, anticonvulsants including gabapentin and pregabalin have largely replaced the other neuropathic pain attenuating drugs [91011]. Carbamazepine has been used in the management of trigeminal neuralgia. Nevertheless, there is a need to identify more efficacious neuropathic pain attenuating drugs.
Many intensive studies have been conducted previously to explore the myriad targets that can be used to ameliorate the anomalies associated with neuropathic pain. Most of the studies have targeted to inhibit noradrenaline and 5-HT transporters [12], voltage-gated sodium channels (Na(v)1.3, 1.7, 1.8, 1.9) [13], voltage-gated calcium channels (Ca(v)1.2, 2.2, 2.3, 3.1, 3.2, 3.3 and alpha-2-delta-1 subunit) [14], purinergic receptors (P1, P2Y and P2X) [1516], transient receptor potential (TRP) channels (TRPM8, TRPA1; TRPV1) [17], metabotropic (mGluR1- mGluR5) and ionotropic (NMDA and AMPA) glutamate receptors and glial glutamate transport [1819]. Many studies have targeted the opening of voltage-gated potassium channels (KATP [Kir6.2/SUR1, Kir6.2/SUR2], KCNQ [K(v) 7.1-K(v) 7.5] and BKCa) [20], GABA receptors and transporters (GAT-1, GAT-3) [21]. Apart from these, many other targets like MAP kinase (ERK, p38 and JNK MAP kinases) [2223], pro-inflammatory mediators (TNF-α, interleukin-1 and interleukin-6) [24], endocannabinoids (CB1 and CB2 receptors, fatty acid amide hydrolase and transporters) [25], PPAR-γ [26], Na+/Ca2+ exchanger [27], nitric oxide [15], calcitonin gene-related peptide [28] and neuronal nicotinic receptors [29]. The drawback have also been exploited of these targets and lack of effective treatment have forced the scientists to discover new targets that would yield desired results.
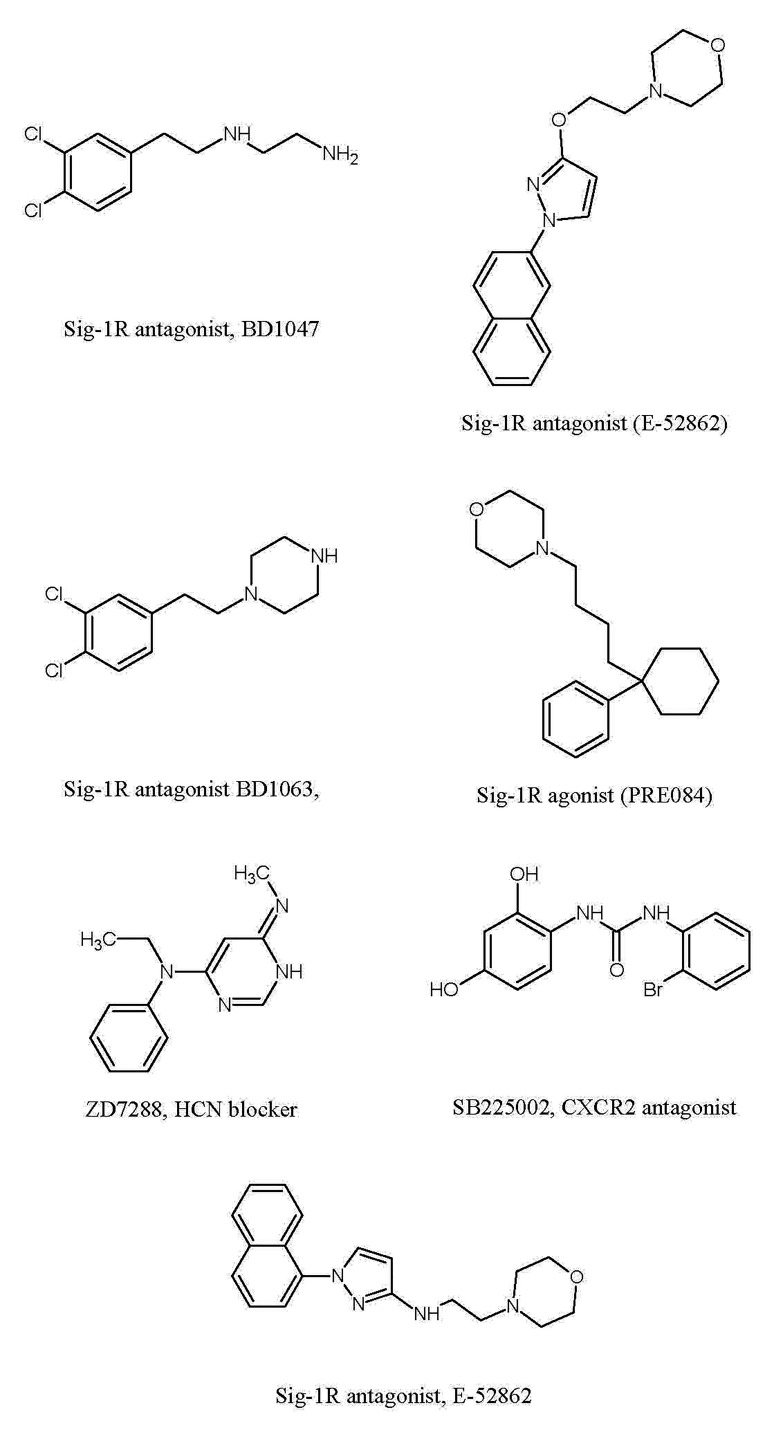
In recent years, more extensive research on neuropathic pain has projected new targets that may potentially attenuate neuropathic pain in diverse models. The targets include D-amino acid oxidase [30], endoplasmic reticulum stress receptors [31], sigma receptors [32], ephrins and Eph receptor tyrosine kinase [33], histone deacetylase [34], HCN channels [35], Cdh-1 [36], Wnt/β-catenin [37], Wnt/Ryk [38] and mitochondrial ATPase [39]. Studies conducted on several animal models of neuropathic pain like chronic constriction injury (CCI), spinal nerve ligation (SNL), partial sciatic nerve ligation (PSNL), spinal cord injury (SCI), spared nerve injury (SNI) have revealed a significant role of these targets in attenuation of neuropathic pain and indicate their effectiveness as therapeutic agents. In this review, these novel targets have been discussed, that can prove to be clinically salubrious and provide better pain management. Specific mechanisms and key roles of these new targets in induction of neuropathic pain have been explained. The exploitation of these targets may provide better understanding of mechanisms underlying neuropathic pain and outlines rationale for developing new strategies to improve pain management.
Go to : 
NEW TARGETS IN NEUROPATHIC PAIN TREATMENT
Sigma receptors
Sigma receptors (Sig-Rs) are unique proteins that were first discovered in 1976 [40] and were originally mischaracterized as a new class of opioid receptors. After sigma receptors were tested and cloned, they were found to have no structural or pharmacological similarity to opioid receptors. Biochemical analysis identified two sub-types of sigma receptors: Sig-1Rs (σ1) and Sig-2Rs (σ2) [41]. Sig-1Rs are located at mitochondria-associated endoplasmic reticulum membrane [42] and are widely distributed in heart, liver, immune cells as well as in the nervous system [43]. Numerous studies indicate the role of Sig-1Rs in pain sensitization [44], and hence, they may be beneficial in the pain management, especially neuropathic pain.
There have been studies showing alterations in the number of Sig-1Rs in the injured and neighbouring neurons in response to nerve injury [45]. The significant role of spinal Sig-1Rs in induction of mechanical allodynia has been demonstrated in CCI model in rats, mainly through the activation of spinal NMDA receptors. Intrathecal administration of Sig-1R antagonist, BD1047 (Fig. 1), twice daily from postoperative days 0 to 5 (induction phase of neuropathic pain) attenuated the mechanical allodynia. On the contrary, administration of BD1047 from days 15 to 20 (maintenance phase) did not modulate mechanical allodynia. These results imply that activation of Sig-1Rs on the dorsal horn neurons may trigger, but do not maintain CCI-induced mechanical allodynia and suggest that Sig-1R antagonists may be useful as a pre-emptive analgesic in chronic pain. The antiallodynic effects of Sig-1R antagonist during the induction phase was correlated with the blockade of expression and phosphorylation of NR1 subunit of NMDA receptor, the most essential component of receptor, in the spinal dorsal horn [46].
The observations from the Sig-1R knockout mice revealed the reduction in wind-up responses (amplification of the nociceptive signals in the spinal cord) in comparison to wild-type mice that had typical wind-up responses in response to sciatic nerve injury. Further, the sensitivity to mechanical and thermal stimuli was significantly attenuated in mice lacking Sig-1Rs. Further, the observations implied that ERK activation was increased in the spinal cords of wild type mice, but no significant changes were observed in Sig-1R knockout mice [47]. Earlier studies had shown that the activation of Sig-1Rs increases the intracellular Ca2+ concentration leading to central sensitization via NMDA receptors [48]. Furthermore, the Ca2+ entry into neurons through NMDA receptors may activate the intracellular ERK signaling [49]. Furthermore, it has been demonstrated that phosphorylation of ERK triggers the development of central sensitization in the spinal dorsal horn neurons to generate and maintain neuropathic pain [5051]. Based on these results, it has been proposed that activation of Sig-1Rs in response to nerve injury may promote Ca2+ dependent ERK phosphorylation to promote pain sensitization.
It is also demonstrated that Sig-1R antagonist (E-52862) (Fig. 1), dose-dependently inhibits capsaicin-induced mechanical hypersensitivity, formalin-evoked nociceptive behaviours (phase I and phase II), and partial sciatic nerve ligation induced mechanical and thermal hypersensitivity. Furthermore, Sig-1R antagonists abolished wind-up phenomenon in isolated spinal cords of mice in response to electrical stimulation of lumbar dorsal horn. Thus, the electrophysiological as well as the pharmacological data support the involvement of these receptors in pain induction [52]. The role of Sig-1Rs in anti-neoplastic drug (paclitaxel)-induced neuropathy has also been explored in mice. Administration of paclitaxel led to development of cold and mechanical allodynia in wild type mice, without any significant effect in Sig-1R knockout mice. Moreover, s.c. administration of Sig-1R antagonist BD1063, before each paclitaxel dose, completely prevented paclitaxel-evoked cold and mechanical allodynia in Wild type mice. Furthermore, there was an increase in the level of pERK1/2 in the dorsal neurons of the spinal cord ten days after paclitaxel treatment in wild type neuropathic mice, whereas, no significant change in pERK1/2 was observed in Sig-1R knockout mice. Therefore, decreased phosphorylation of ERK after sciatic nerve injury or following paclitaxel administration in Sig-1R knockout mice may be responsible for the marked reduction of cold and mechanical allodynia [53].
Pharmacological blockade or genetic knockout of Sig-1Rs reduces the development of atypical axonal mitochondria in the saphenous nerve myelinated fibers and neuropathic pain after paclitaxel administration. Paclitaxel-induced mitochondrial abnormalities may occur via opening of mitochondrial permeability transition protein (mPTP), after its binding to β-tubulin [54], and/or to bcl-2, which could induce mitochondrial swelling and increase the release of mitochondrial Ca2+ to the cytoplasm [55]. Therefore, Sig-1Rs may possibly modulate bcl-2 proteins to indirectly regulate mPTP opening and mitochondrial swelling, thus, implying the potential role of Sig-1R antagonists in prevention of paclitaxel-induced neuropathic pain [32].
In a CCI model, activation of spinal Sig-1Rs is shown to activate p38 mitogen-activated protein kinase (p38MAPK), which plays a vital role in many chronic pain states including mechanical allodynia [56]. However, early i.t. treatment with the Sig-1R antagonist (BD1047) during the induction phase prevented the increase in p38 MAPK expression in spinal dorsal horn neurons, and attenuated mechanical allodynia [57]. Sig-1Rs have been reported to modulate intracellular Ca2+ signaling and activate Ca2+ dependent enzymes, leading to p38 MAPK activation in the dorsal horn neurons [58]. The facilitatory effects of Sig-1R agonists in pain perception are also mediated through nitric oxide (NO) signaling via neuronal nitric oxide (nNOS) activation [59], which is recognized to be closely associated to p38 MAPK signaling. These findings together indicate that Sig-1Rs can modulate the phosphorylation of p38 MAPK via Ca2+ dependent cascade and/or NO signaling pathway, which is vital for induction of mechanical allodynia.
Intra-hippocampal injections of Sig-1R antagonist, BD1063 (5–50 µg/rat), and Sig-2R antagonist, SM21 (1–5 g/rat) is shown to significantly attenuate agmatine-induced enhancement in thermal hyperalgesia, cold allodynia, mechanical allodynia and TNF-alpha levels. Since, the overproduction of TNF-alpha is associated with development of neuropathic pain [6061], therefore, it was proposed that Sig-1R and Sig-2R antagonists may attenuate pain by reducing TNF-alpha levels in hippocampus in nerve ligated animals [62].
It has been shown that intrathecal administration of Sig-1R agonist (PRE084) in mice and CCI in rats increases the reactive oxygen species production through NADPH oxidase 2 (Nox2) in the lumbar spinal cord dorsal horn to produce mechanical allodynia and thermal hyperalgesia. Furthermore, intrathecal administration of Sig-1R antagonist (BD1047) (Fig. 1) reduced CCI-induced Nox2 activation (reactive oxygen species generation) and pain behaviour [63] suggesting that sigma receptors may activate free radical production to induce pain. This hypothesis was further supported by the finding showing that the Sig-1R agonists increase the p47 phox expression (the most critical component responsible for Nox2 activation) and Sig-1R antagonist (BD1047) suppresses the p47 phox expression in CCI rats.
The anti-nociceptive actions of selective Sig-1R antagonist has also been linked with the modulation of three key neurotransmitters i.e., glutamate, GABA and nor-adrenaline, in the spinal dorsal horn. Systemic administration of Sig-1R antagonist attenuated formalin-induced flinching and lifting/licking in phase I (initial acute pain phase) and phase II (prolonged tonic pain phase), indicating both spinal and supra-spinal actions. Furthermore, it also attenuated formalin-induced increase in glutamate and decrease in nor-adrenaline levels in the dorsal horns of spinal cord, without altering the GABA levels. Accordingly, it is proposed that Sig-1R antagonists may modulate intracellular Ca2+ ions to facilitate nor-adrenaline release, which may activate spinal alpha2 receptors to attenuate the release of glutamate, leading to their antinociception effects in formalin model [64].
The role of D-serine in the induction of neuropathic pain through activation of Sig-1R in CCI model of neuropathic pain has been explored in mice. After nerve injury, there was an increase in the levels of D-serine and serine-racemase (SR) in astrocytes of the superficial dorsal horn. It is found that SR coexists in the Sig-1Rs expressing astrocytes. Intrathecal administration of BD-1047 attenuated CCI-induced increase in D-serine and SR, and blocked the induction of mechanical allodynia. Moreover, selective blockade of SR by L-serine O-sulfate potassium salt (LSOS) inhibited the development of mechanical allodynia, but not thermal hyperalgesia in CCI mice. Therefore, it was proposed that Sig-1Rs activation was responsible for the increased expressions of D-serine and SR in the astrocytes, affecting dorsal horn neurons that are involved in induction of mechanical allodynia in neuropathic pain [65].
From these extensive sigma receptor-based studies, it may be concluded that Sig-1Rs play a significant role in the induction and maintenance of neuropathic pain through different mechanisms. Sig-1Rs modulate the intracellular Ca2+ entry that leads to the phosphorylation of ERK, NMDA, p38 MAPK and activation of NO signaling, all of which forms the basis for induction of neuropathic pain. Moreover, activation of Nox2 and TNF-alpha through Sig-1Rs, also play a major role in the induction of neuropathic pain by production of reactive oxygen species. Therefore, blocking of Sig-1Rs may result in attenuation of neuropathic pain (Fig. 2).
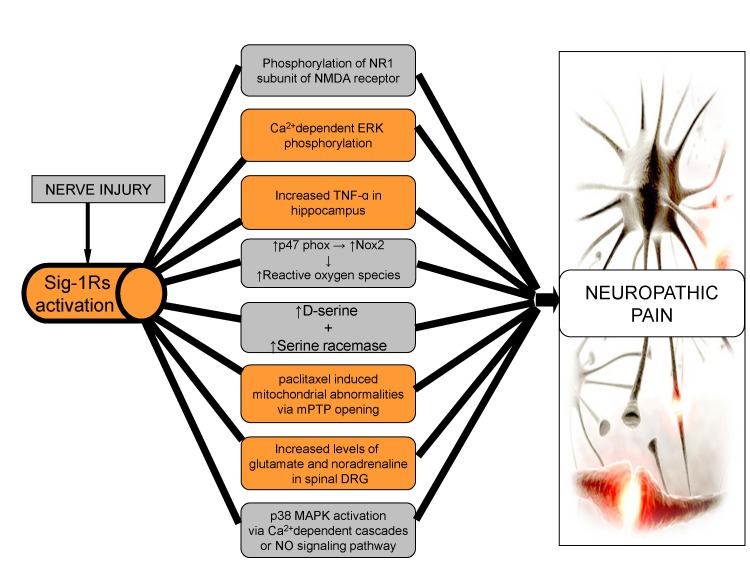 | Fig. 2Role of sigma receptors in neuropathic pain.After nerve injury, there is activation of Sig-1Rs, with an increase the intracellular entry of Ca2+, resulting in increased phosphorylation of NMDA, ERK, p38 MAPK and activation of NO signaling leading to neuropathic pain. Increased levels of D-serine, glutamate and nor-adrenaline along with mitochondrial abnormalities via Sig-1Rs are major factors in the induction of neuropathic pain. Production of reactive oxygen species through Nox2 and TNF-alpha, via Sig-1Rs may also contribute to neuropathic pain.
|
Ephrins and Eph receptor tyrosine kinase
Eph receptor tyrosine kinases and their ligands, ephrins are involved in myriad of developmental aspects, including tissue patterning, angiogenesis, axon guidance and synapse formation [66]. Several Eph and ephrin receptor proteins are expressed in the laminae I-III of spinal dorsal horn and on small and medium sized DRG neurons [67]. EphB receptors and ephrins modulate synaptic activity in the spinal cord, contributing to sensory abnormalities in persistent pain states in NMDA dependent manner [6869], therefore, suggesting a key role of ephrin and Eph system in physiologic and pathologic pain modulation in the spinal cord level.
L5 spinal nerve injury increases the expression of ephrin B2 (in the neurons of DRG and spinal cord) and Eph B1 isoforms in the neurons of spinal cord. Administration of ephrinB2 siRNA led to reduction in the expression of ephrinB2 and attenuated nerve injury-induced mechanical allodynia. Hence, it may be implied that activation of EphB1/ephrin B2 signaling pathway contribute in neuropathic pain [70]. There is a correlation between development of hyperalgesia and increase in the expression of ephrinB1 and EphB receptor proteins in DRG neurons, dorsal horn and IB4 positive nociceptive terminals in CCI and dorsal rhizotomoy (DR) model. After nerve injury the activation and redistribution of ephrinB-EphB in DRG and dorsal horn contribute to neuropathic pain. Intrathecal administration of EphB-receptor antagonists (EphB1-Fc and EphB2-Fc chimeras) was shown to suppress the induction and maintenance of nerve injury-induced mechanical allodynia and thermal hyperalgesia. Moreover, EphB antagonists also prevented nerve injury-induced hyper-excitability of nociceptive small DRG neurons, sensitization of dorsal horns neurons and LTP of synapses between C fibers and dorsal horn neurons. Furthermore, intrathecal injection of EphB-receptor activators (ephrinB1-Fc and ephrinB2-Fc), in uninjured animals, induced the thermal hypersensitivity and reduced the threshold for LTP. Accordingly, the up-regulation of ephrinB1 and EphB1 receptor proteins after nerve injury may increase the excitability of nociceptive related neurons and synaptic plasticity at spinal level, leading to induction of neuropathic pain [71].
The ephrin-B2 conditional knockout (CKO) mice, with deletion of ephrin-B2 from Nav 1.8-expressing nociceptors, demonstrated the reduction in pain behavior in different models of pain. Furthermore, there was significant decrease in tyrosine phosphorylation of NMDA receptor subunit NR2B in the dorsal horn suggesting that ephrin-B2 signaling plays a vital role in regulating pain thresholds, with its mechanisms associated with NMDA receptor phosphorylation [72]. It is demonstrated that the activation of MAPKs modulates the spinal nociceptive information through ephrinBs/EphBs signaling in CCI and formalin induced mice models of neuropathic pain. Intrathecal injection of ephrinB1-Fc produced a dose-dependent thermal hyperalgesia and mechanical allodynia, along with an increase in the expression of spinal c-Fos and p-MAPKs. The activated forms of p38 and JNK were localized in the spinal neurons and astrocytes, while p-ERK was localized in the spinal neurons. Inhibition of spinal MAPK was observed to reverse the pain behavior and prevent ephrinB1-Fc-induced hyperalgesia and spinal Fos protein expression. The blockade of EphB receptors alleviated formalin and CCI induced pain along with attenuation of spinal MAPKs and c-Fos protein activation Moreover, pre-treatment with NMDA receptor antagonist (MK-801), suppressed ephrinB1-Fc induced hyperalgesia and activation of spinal MAPKs. Therefore, it can be implied that ephrinsB1-Fc leads to activation of MAPK pathways through NMDA receptors to induce pain behavior [73].
The role of phosphatidylinositol 3-kinase (PI3K), as a downstream effector in the modulation of spinal nociceptive information, in relation to ephrinBs and EphBs in formalin and CCI-induced inflammation and neuropathic pain has been shown in mice. Intrathecal injection of ephrinB1-Fc is shown to induce mechanical allodynia and thermal hyperalgesia, increase spinal c-Fos and PI3Kp110γ expression and phosphorylate AKT (p-AKT), a downstream target of PI3K. Observations suggested that pre-treatment with wortmannin, a PI3K inhibitor, prevented ephrinB1-Fc induced activation of AKT, spinal c-Fos protein expression and reversed the pain behaviors. Similarly, blockade of spinal EphBs receptors suppressed formalin and CCI-induced inflammation and neuropathic pain behaviors by reducing the levels of spinal PI3K, c-Fos expression and reducing the phosphorylation of AKT. Therefore, it can be implied that PI3K has a significant role in modulation of nociceptive information related to ephrinsBs and EphBs via ERKs and EphBs via ERK activation [74].
The role of protein kinase C-γ (PKCγ) as a downstream effector in regulation of spinal pain processing via ephrinB-EphB signaling has been shown in various mice models of neuropathic pain. Observations indicated that intrathecal injection of EphB receptor activator (ephrinB2-Fc) induced mechanical allodynia and thermal hyperalgesia, along with the increase in the expression of spinal PKCγ. In spinal PKCγ knockout mice, there was no ephrinB2-Fc induced pain behavior. Moreover, intrathecal injection of EphB receptor blocker (EphB2-Fc) led to suppression of formalin-induced inflammatory, CCI induced neuropathic pain, tumor cell implantation (TCI)-induced bone cancer pain behaviours. Moreover it also decreased the activation of spinal PKCγ. Intrathecal injection of NMDA receptor blocker, (MK801), also suppressed ephrinB2-Fc-induced spinal PKCγ activation and pain behavior, confirming the key role of PKCγ in spinal pain processing through ephrinB-EphB signaling [75].
From above studies, it can be concluded that an increase in EphrinBs/EphBs signaling leads to increase in activation of PKCγ, MAPK, c-Fos, p-AKT, P13K and phosphorylation of NMDA. All these factors are responsible for increased excitability of nociceptive neurons and synaptic plasticity, which are fundamental mechanisms leading to induction of neuropathic pain (Fig. 3).
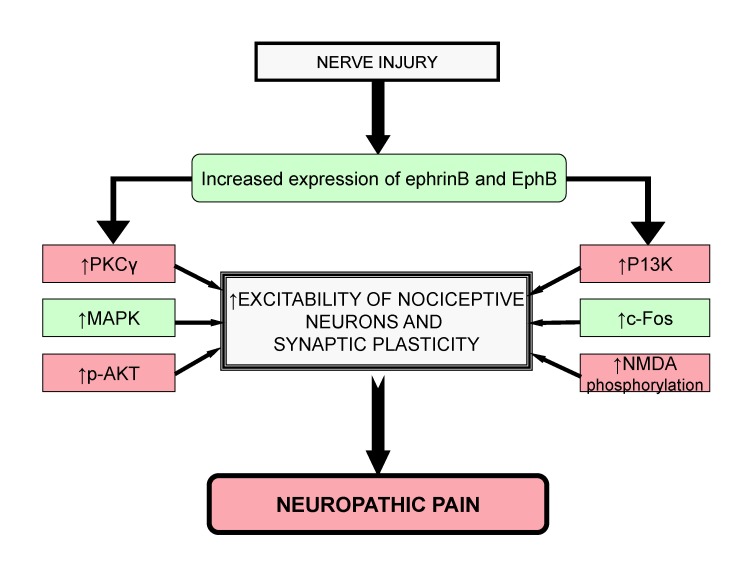 | Fig. 3Role of Ephrins and EphB receptor tyrosine kinase leads in neuropathic pain.After nerve injury, the increased levels of PKCγ, MAPK, c-Fos, p-AKT, P13K and NMDA phosphorylation through ephrinB and EphB signaling lead to increased excitability of nociceptive neurons and synaptic plasticity that contributes to neuropathic pain.
|
Endoplasmic reticulum stress (ERS) receptors
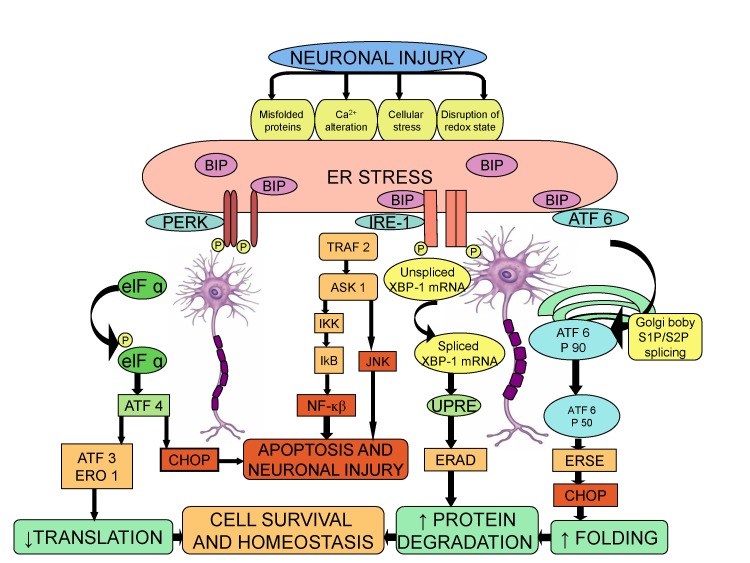
In a number of pathological conditions like glucose deprivation, depletion of calcium stores, exposure to free radicals and accumulation of unfolded proteins, there is disruption of the proper functioning of ER and thereby, causing ER stress, which severely impairs the protein folding [7677]. The cells react to ER stress by increasing the expression of molecular chaperone such as Binding Immunoglobulin Proteins BIP (GRP 78), which initiates a defensive process called Unfold Protein Response (UPR) [78]. The transcriptional activation of BIP is used as a marker of UPR initiation [77]. The UPR is mediated by three ER stress receptors: PKR-like ER kinase (PERK), inositol-requiring enzyme 1 (IRE1), and the activating transcription factor-6 (ATF6) [79]. Studies suggest that ER stress response is a key reaction in inflammatory diseases, including neuroinflammation [80]. The signaling pathway between ER stress and neuroinflammatory response is connected through several mechanisms, including reactive oxygen species, calcium, nuclear factor-κB (NK-κB), and mitogen-activated protein kinase (MAPK) [81]. The pro-inflammatory substances trigger the ER stress response, which in turn may contribute in the development of neuropathic pain [8283].
The role of ER stress response in neuropathic pain has been demonstrated in a Complete Freund's Adjuvant (CFA) injected rat model of orofacial inflammatory pain. The protein and mRNA levels of ER stress response genes, BIP (GRP78), p-eIF2α and ATF4 were significantly increased in the trigeminal ganglion (TG). The expression of BIP (GRP78) was increased in all sized TG neurons, while the p-eIF2α expression was increased in small and medium sized neurons. Administration of ER stress modulator, salubrinal, into TG for 5 days following CFA injection, attenuated heat hyperalgesia suggesting that induction of ER stress response in the primary sensory neurons is responsible for orofacial inflammation and pain [84]. The role of ER stress and UPR was examined in L5 spinal nerve ligation (SNL)-induced mirror-image neuropathic pain in rats. Nerve ligation led to activation of BIP (GRP 78) followed by activation of IRE-XBP1 and ATF6 pathways, but not PERK-e1F2 pathway. Accordingly, the increased levels of BIP in the dorsal horn indicate the induction of ER stress in response to neuronal injury. The increased level of spliced XBP-1 in the dorsal horn is a result of activation of IRE-1, as activation of IRE-1 induces the splicing of XBP-1 mRNA via cleavage of its intron [85]. Furthermore, it is also well documented that the spliced XBP-1 functions as a transcription factor for ER stress related genes and may be critical in initiating the ER stress response [86]. The activation of ATF6 pathway is mediated through BIP, which cleaves it from ER membrane and translocates it to the Golgi complex, followed by the transcription of UPR target genes by the cleaved 50-kDa product of ATF-6 fragment [87]. The intrathecal injection of siRNA for ATF-6 attenuated the neuropathic pain, again emphasizing the cardinal role of ER stress in the induction of neuropathic pain [83].
The role of epoxy fatty acids (EpFAs) and ER stress-regulated response in diabetes-induced neuropathic pain has been investigated. Since, soluble epoxide hydrolase (sEH) degrades EpFAs, the natural analgesic mediators; therefore, blocking of sEH produces analgesic actions. The precise mechanisms involved in analgesic effects of EpFAs are unknown; however, it has been postulated that naloxone antagonizes analgesic effects of these fatty acids suggesting the possible role of opioids in producing anti nociceptive effects. Inhibitors of sEH reduced ER stress markers and neuropathic pain and it also suggested that EpFAs are upstream modulators of ER stress pathways [88].
Taken together, the results imply a major role of ER stress pathways in regulating the excitability of nociceptive system and inflammatory pain in peripheral nervous system. Although, during cellular stress the main goal of UPR pathway is to promote cell survival, yet, the continuous and persistent activation of ER stress response may render the spinal neurons vulnerable to neuronal injury in response to neuropathic pain stimuli (Fig. 4).
β-Catenin, Wnt/Ryk and Wnt/β-catenin
β-catenin is a subunit of cadherin protein complex, localized to synaptic contacts, and is associated with synapse formation and neuronal circuit assembly during development [89]. Wnts belong to the family of glycoproteins and play a major role in many aspects of CNS development such as neural induction, cell proliferation, neuronal migration, axon guidance, dendritic arborization and synaptogenesis [90]. Ryk, a trans-membrane protein receptor of tyrosine kinase, is required for normal axon guidance and neuronal differentiation [91] and it binds with Wnt to modulate its activity [92]. Menin (MEN I), a transcription factor product of multiple endocrine neoplasia type I, is a tumor suppressor gene [93] that is localized to the nucleus and is also critical mediator of synapse formation between neurons. Moreover, menin has an essential role in Wnt/β-catenin signaling through histone methylation of downstream target gene promoters [94]. The Wnt/β-catenin pathway, a canonical Wnt signaling pathway, has a significant role in neural development, axonal guidance, neuropathic pain remission and neuronal survival [95]. β-catenin, Wnt/Ryk and Wnt/β-catenin pathways are explored as a therapeutic targets for treating neuropathic pain.
There is an increase in the expression of β-catenin and menin in the ipsilateral spinal dorsal horn of rats in response to spared nerve injury (SNI), which is associated with development of mechanical hypersensitivity. It is possible that menin may induce the formation of new synapses and sprout Aβ-fibers from lamina III to lamina II of dorsal horn and these changes may contribute in the induction of neuropathic pain [96]. An increase in expression of Wnt3a, a prototypic Wnt ligand of Wnt/β-catenin pathway, in the dorsal horn of spinal cord after partial sciatic nerve ligation (PSL) has also been reported. Intrathecal administration of XAV939, a Wnt/β-catenin signaling inhibitor, effectively attenuated the induction of neuropathic pain and microglial activation. Furthermore, intrathecal treatment with Wnt3a in the lumbar spinal regions of naive animals stimulated the development of allodynia and triggered the release of brain-derived neuropathic factor (BDNF) from microglial cells. Hence, it may be possible that up-regulation of Wnt3a in the dorsal horn activates the microglial cells to secrete BDNF, which is responsible for neuropathic pain induction [97].
An increase in the expression of Wnts in the DRG and spinal cord and activation of Wnt/Frizzled/β-catenin signaling in the spinal cord in CCI model and tumor cell implantation (TCI)-induced bone cancer in mice model has been reported. Wnt signaling enhanced the production of pro-inflammatory cytokines, which led to induction of neuropathic pain. It was found that Wnt inhibitors attenuate mechanical allodynia and thermal hyperalgesia in neuropathic pain models suggesting the critical role of Wnts in induction of neuropathic pain [88]. An increase in expression of Wnt3a, Wnt5b and Ryk receptors in the primary sensory neurons, dorsal horn neurons and astrocytes has been observed after sciatic nerve injury. Inhibition of Wnt/Ryk receptor signaling led to blockade of neuropathic pain without any significant effect on normal pain sensitivity and locomotor activity. In vivo and in vitro blockade of Ryk receptors with anti-Ryk antibody suppressed nerve injury-induced increase in intracellular Ca2+, sensory neuronal hyper-excitability, enhanced plasticity of synapses between afferent C-fibers and dorsal horn neurons, implying that Wnt/Ryk signaling may be a key mechanism underlying the pathogenesis of neuropathic pain [98].
The neuroprotective mechanism of rapamycin in spinal cord injury in rats has been correlated with an increase in expression levels of β-catenin and BDNF. Accordingly, it may be proposed that rapamycin reduces neuronal injury by inhibiting the apoptosis of neurons, and increasing the expression of BDNF, via activation of Wnt/β-catenin pathway [90]. It has been shown that nerve injury increases the levels of TCF4, β-catenin and GSK-3β within the neurons of DRG and spinal cord. Furthermore, intrathecal injection of Wnt/β-catenin inhibitor (IWR-1-endo) and TCF4 small interfering RNA (siRNA) attenuated CCI-induced mechanical allodynia and thermal hyperalgesia and reduced the expression of β-catenin and GSK-3β. Thus, it may be suggested that TCF4 in DRG and spinal cord may be involved in maintenance of CCI-induced neuropathic pain through Wnt/β-catenin signaling pathway [99].
Based on these studies, it may be proposed that Wnt/β-catenin along with Wnt/ryk signaling plays a critical role in induction of neuropathic pain, either through production of pro-inflammatory cytokines or through secretion of BDNF from the microglial cells. The binding of Wnt to its receptor suppresses the GSK-3β phosphorylation of β-catenin, which on accumulation binds to TCF4 and leads to activation of Wnt target genes, causing neuropathic pain (Fig. 5).
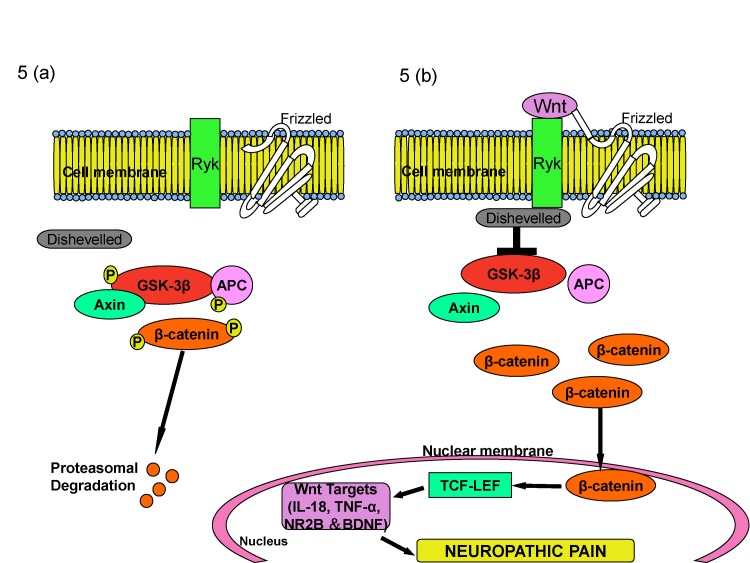 | Fig. 5Role of β-catenin system in induction of neuropathic pain.In the physiological state, absence of Wnt leads to decreased levels of β-catenin in the cytoplasm as GSK-3b phosphorylates b-catenin and leads to its proteosomal degradation (A). During the nerve injury, Wnt binds to its receptor frizzled and suppresses the GSK-3β phosphorylation of β-catenin. Increased β-catenin binds to TCF4 and activates Wnt target genes that are responsible for neuropathic pain (B).
|
Hyperpolarization-activated cyclic nucleotide-gated cation channels (HCN)
Hyperpolarization-activated and cyclic nucleotide-gated (HCN) channels are non-selective, ligand-gated cation channels [100] and are present in the cells of heart and central nervous system [101]. HCN channels are encoded by four genes (HCN1, HCN2, HCN3, HCN4), and the activation of these channels generates inward currents during hyperpolarization membrane potential. The physiological functions of these channels include normalization of synaptic inputs, regulation of resting membrane potential and modulation of intrinsic cellular frequency characteristics [102]. Various studies conducted on HCN channels imply their significant role in induction of neuropathic pain [103104].
It is reported that the spinal HCN channels at the primary afferent terminals contribute to chronic pain in formalin and SNL-induced neuropathic pain models in mice. Intra-peritoneal and intrathecal administration of eugenol reversed peripheral nerve injury induced mechanical allodynia in trigeminal neurons by inhibition of HCN channels. Eugenol attenuates CCI of infraorbital nerve-induced mechanical allodynia and inhibits HCN current with IC50 of 157 µM in trigeminal ganglion neurons. Furthermore, eugenol induced inhibition of HCN current was diminished by increase in intracellular cAMP levels [104].
It has been reported that the genetic deletion of HCN2 diminishes cAMP sensitive component causing inward current and suppresses the action potential firing caused by elevation of cAMP levels in nociceptors. The deletion of HCN2 specifically in Nav 1.8 expressing nociceptors showed normal pain threshold in neuropathic pain mice, indicating a vital role of HCN2-driven action potential firing expressed in Nav 1.8 nociceptors [103]. The impact of changes to ventral-lateral periaqueductal gray (vIPAG) HCN channels on the neural activity in CCI model of neuropathic pain has also been reported. An increase in expression of HCN1 and HCN2 channels, augmentation of hyperpolarization-activated current amplitudes to more positive potentials and increase in action potential firing following CCI has been reported. Forskolin, which elevates intracellular cAMP, mimics CCI-induced changes in neuronal excitability in vIPAG. Furthermore, ZD7288, a HCN blocker, attenuated spontaneous EPSCs and reversed the effects of forskolin [105].
Ivabradine, an anti-anginal agent, attenuates pain in different models of inflammatory and neuropathic pain through HCN ion channels. Ivabradine blocks HCN channels, expressed on the small sensory neurons, and suppresses the action potential firing in nociceptive neurons by increasing the intracellular cAMP [106]. A significant increase in HCN2-immunoreactivity in small (<30 µm) DRG neurons (specifically in IB-4 negative neurons) has been observed in L5 spinal nerve axotomy model in rats. Furthermore, intra-plantar treatment with ZD7288 (100 µM) was shown to decrease mechanical pain behavior [107]. Pulsed Radio-Frequency treatment (PRF) is shown to decrease thermal hyperalgesia, mechanical allodynia and mechanical hyperalgesia along with an increase in the levels of HCN1 and HCN2 in DRG in CCI model [97].
The dysfunction of HCN channels in the pyramidal neurons of the medial Pre-frontal Cortex (mPFC) has also been reported in SNI model in rats. Hyper-polarizing shift in the SNI neurons was observed in the II/III pyramidal neurons of the contralateral mPFC. Furthermore, it was found that hyperpolarization shift was abolished by intracellular treatment with bromo-cAMP. Acute HCN blockade by local injection of ZD7288 in mPFC of SNI rats suppressed cold allodynia indicating that changes in the cAMP/Protein kinase A axis in mPFC neurons may be responsible for alterations in HCN channel function, which may further influence the descending inhibition of pain pathways in neuropathic pain [108].
Thus, it may be concluded that in response to nerve injury, an increase in intracellular cAMP level leads to action potential firing that increases the expression of HCN in DRG neurons and an increase in HCN expression is responsible for the induction of neuropathic pain.
D-Amino acid oxidase (DAAO)
Studies conducted in animal models of neuropathic pain have suggested that spinal DAAO, restricted within the astrocytes [109110], plays a pronociceptive role and might be a potential target for treatment of neuropathic pain [72]. DAAO is a peroxisomal enzyme that catalyzes the oxidative deamination of polar and neutral D-amino acids to α-keto acids, ammonia and hydrogen peroxide [109]. In the CNS, D-amino acid oxidase is confined to the lower brain stem, cerebellum and spinal cord, with lower levels present in the mid brain, cortex and hippocampus [111].
The potential role of DAAO in neuropathic pain in a rat model of L5/L6 spinal nerve ligation has been investigated. Nerve ligation led to increase in mRNA expression and enzyme activity of DAAO in the lumbar spinal cord. The systemic injection of DAAO inhibitor, sodium benzoate, specifically blocked neuropathic pain by blocking the DAAO activity in the lumbar enlargement of spinal cord. Direct intrathecal injection of sodium benzoate (30 µg/rat) specifically reduced spinal nerve ligation-induced neuropathic pain. Furthermore, systemic and intrathecal administration of sodium benzoate blocked formalin-induced hyperalgesia and spinal nerve ligation induced neuropathic pain to an equal extent [72]. Study using small interference RNA (siRNA) has suggested that the down regulation of spinal DAAO expression and enzymatic activity leads to analgesia with its mechanism potentially related to inhibition of astrocytes in the spinal cord. Multiple daily intrathecal injections of siRNA for DAAO (30 µl/day) markedly inhibited spinal DAAO gene and protein expressions, DAAO enzymatic activity, expression of glial fibrillary acidic protein (GFAP), a biomarker of astrocytes activation and formalin-induced tonic phase pain in rats [30]. In another study, DAAO inhibitor, 5-chloro-benzo[d]isoxazol-3-ol (CBIO) was shown to block formalin-induced tonic phase pain and spinal hydrogen peroxide levels, suggesting that spinal DAAO produces algesia via production of hydrogen peroxide [112]. Hydrogen peroxide, most likely derived from the spinal astrocytes [113], is an active and stable reactive oxygen species that is produced form the oxidation of D-amino acids by DAAO [110113], Hydrogen peroxide may be involved in induction of pain either directly or indirectly through N-methyl-D-aspartate (NMDA) receptor activation [98].
Therefore, it is plausible to suggest that nerve injury or toxic chemical may increase the expression as well as the activity of DAAO in the spinal cord to activate the central astrocytes, which may increase the production of reactive oxygen species. The latter in turn directly or indirectly through increase in phosphorylation of NMDA receptors may induce neuropathic pain (Fig. 6).
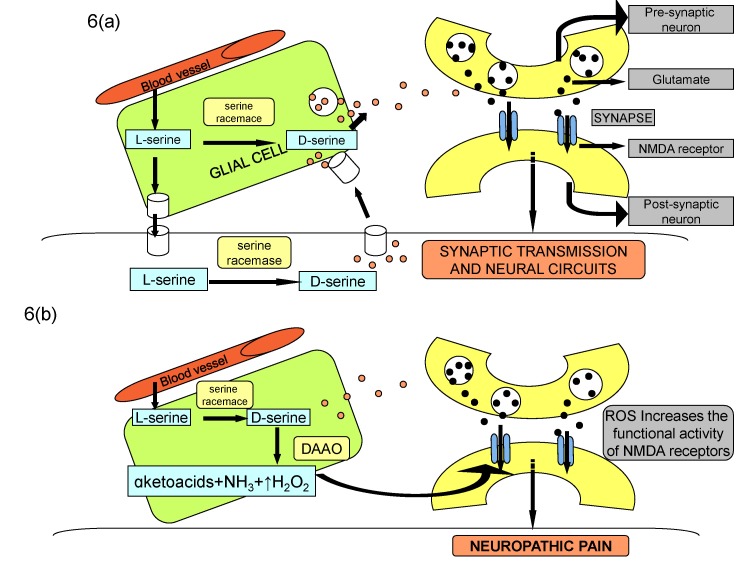 | Fig. 6NMDA-mediated synaptic transmission during physiological state (A) and nerve injury state (B).Binding of glutamate and co-agonist D-serine to NMDA receptor subunit modulates synaptic transmission and plasticity in normal state. Increase in DAAO activity after nerve injury decreases the levels of D-serine, which may indirectly increase the synthesis of reactive oxygen species to induce neuropathic pain through increased NMDA phosphorylation and consequent enhanced synaptic plasticity.
|
Histone deacetylase
Histone deacetylase enzymes (HDAC) along with histone acetyl-transferases (HATs) control the process of acetylation of histone lysine residues, such as lysine residue9 (H3K9ac), which further modulates the expression of transcription factors [114] and also promotes transcriptional elongation. Histone acetylation is one of the epigenetic mechanisms that is hypothesized to be involved in induction of neuropathic pain, with its causes primarily rooted in altered transcriptional expression. There has been an increase the expression of histone deacetylase enzymes following nerve injury or inflammatory conditions that induce deacetylation of histone proteins and histone deacetylase inhibitors attenuate neuropathic pain in neuropathic and inflammatory pain [34].
There is an increase in expression of histone deacetylase 4 in response to spinal nerve ligation in rats and inhibition of histone deacetylase 4, using LMK 235, prevented development of allodynia [115]. In spinal nerve ligation model, an increase in histone deacetylase 1 and decrease in histone (H3) acetylation has been documented. Treatment with baicalin (an anti-inflammatory flavonoid) significantly attenuated deacetylase 1 expression and reversed pain sensitivity [116]. A study from our laboratory documented neuropathic pain attenuating potential of histone deacetylase inhibitor (sodium butyrate) in CCI model in rats [117]. Administration of another histone deacetylase inhibitor (trichostatin A) has been shown to suppress inflammatory pain symptoms [112]. There have been other studies showing that histone deacetylase inhibitors such as valproic acid [118], MS-275 and suberoylanilide hydroxamic acid reduce the nociceptive response [119]. Administration of curcumin has been shown to attenuate pain in formalin model along with hypo-acetylation of histones H3 and H4 in dorsal root ganglia. Furthermore, pretreatment with histone deacetylase inhibitor, suberoylanilide hydroxamic acid, enhanced the analgesic activity of glutamate agonist (LY379268) proposing the potential of histone acetylation to attenuate pain [120]. A recent study has also shown an increase in expression of histone deacetylase 1 in microglia and astrocytes in the dorsal horn neurons in response to nerve injury suggesting that increase in histone deacetylase in the astrocytes may be a key factor in inducing pain [121]. Another study has shown an increase in expression of histone deacetylase 2 specifically in astrocytes, with no significant change in neurons of dorsal horn in nerve injury model. The mechanisms responsible for histone hypoacetylation-induced pain include decrease in metabotropic glutamate receptors in the DRG and spinal cord [120]; increase in cytokines [117]; decrease in glutamate transporters [118]; down-regulation of mu-opioid receptors [122] and decrease in glutamic acid decarboxylase 65 expression, leading to decrease in synaptic GABAergic inhibition [123].
Therefore, it may be proposed that nerve injury produces epigenetic changes due to activation of histone deacetylase enzymes, which may decrease histone acetylation to induce pain and histone deacetylase inhibitors may be employed to inhibit neuropathic pain (Fig. 7).
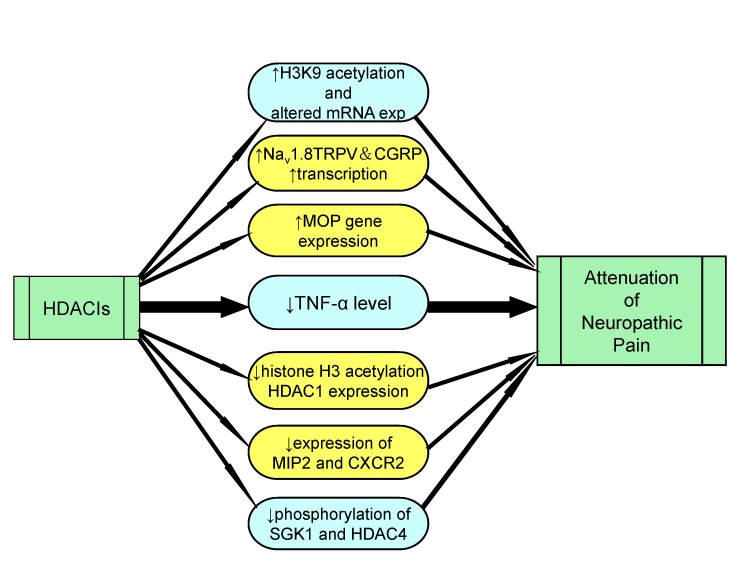 | Fig. 7Role of HDAC inhibitors in attenuation of neuropathic pain.Histone deacetylase inhibitors helps in management of neuropathic pain by increasing the acetylation of H3K9, transcription of Nav 1.8, TRPV and CGRP and increasing expression of MOP genes. HDACIs also result in decreased production of inflammatory mediators, reduction of histone H3 acetylation and reduced phosphorylation of SGK1 and HDAC4.
|
Mitochondrial ATPase
Mitochondria, a powerhouse of cell, play a major role in energy production, cellular signaling, apoptosis, ROS production, and calcium homeostasis [124]. Both mitochondrial and bioenergetic dysfunction is assumed to play a significant role in induction of neuropathic pain [39]. It has been shown that chemotherapeutic agents like paclitaxel and oxaliplatin lead to nitration of superoxide anion in the mitochondria to form peroxynitrite. It may be possible that during nerve injury, formation of peroxynitrite may damage the mitochondria and decrease the production of ATP, which may contribute in induction of neuropathic pain [125]. Another study demonstrates the role of mitochondrial ATP synthase in DRG after sciatic nerve injury in adult rats. Immunohistochemical and Western Blotting studies showed the decreased expression of mitochondrial ATP synthase in L4-L5 DRG neurons in the un-myelinated C-fibers and thinly myelinated Aδ fibers, suggesting the decreased ATP production. The most cardinal observation was that the intrathecal ATP injection reduced thermal and mechanical hyperalgesia after nerve injury [126]. It is important to understand that ATP is not an energy product; rather it may also act on specific membrane receptors to modulate pain transmission [127].
It has been shown that oxygen consumption, which is vital for ATP synthase activity is significantly reduced in injured nerves in response to partial sciatic nerve ligation (PSNL) in mice. Nerve injury led to increase in the number of immune cells that contributed to neuroinflammation and increased the oxygen demand. Therefore, the development of hypoxia (due to increased oxygen demand) further reduced the ability of mitochondria to carry out oxidative phosphorylation, which may be responsible for decrease in the maximal respiratory capacity of injured nerves. An increase in the number of immune cells was also responsible for increased glycolysis and lactic acidosis, which lead to development of tissue acidosis [39]. Tissue acidosis has been known to cause constant ongoing pain [128].
Therefore, it is concluded that after nerve injury, the increased oxygen consumption and glycolysis leads to hypoxia and acidosis, which trigger other anomalies responsible for mitochondrial and bioenergetic dysfunction. The decreased energy production associated with mitochondrial dysfunction reduces Na+/K+ATPase activity, which is responsible for membrane hyper-excitability. It is thus, vital to protect mitochondria to ameliorate the symptoms of neuropathic pain (Fig. 8).
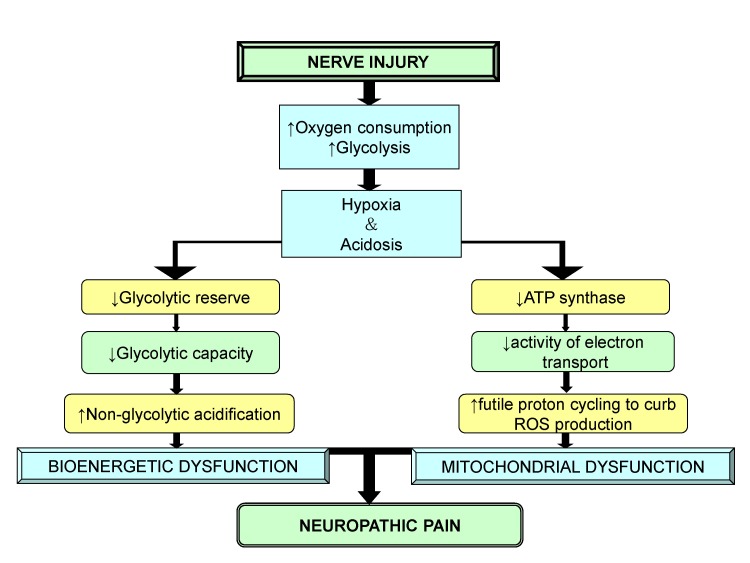 | Fig. 8Role of mitochondrial and bioenergetic dysfunction in neuropathic pain.After nerve injury, an increase in oxygen consumption result in hypoxia, acidosis, decreased glycolytic reserve and decrease in energy production that leads to mitochondrial and bioenergetic dysfunction causing neuropathic pain.
|
Cdh-1, a co-activator subunit of anaphase-promoting complex/cyclosome (APC/C)
Cdh-1 is a tumor suppressor gene that belongs to cadherin super family [129]. It leads to production of calcium-dependent cell-cell adhesion glycoprotein and several reports indicate the role of Cdh-1 in induction of neuropathic pain [130]. Anaphase-promoting complex/cyclosome (APC/C) and Cdh-1 are linked to various neurobiological functions, and the most vital is the regulation of synaptic differentiation and transmission [131]. It has been reported that APC/C-Cdh1 mediates synaptic plasticity through an EphA4-dependent signaling pathway in cortical neurons [132] and in anterior cingulate cortex (ACC), that is a major cortical area involved in processing pain related emotion and transmission of pain sensation [133]. In spared nerve injury (SNI) model, up-regulation of c-Fos in ACC was demonstrated suggesting the induction of central sensitization. A significant decrease in nuclear export of Cdh-1 after SNI was also observed, suggesting the decreased APC/C-Cdh1 activity in the ACC. The decreased APC/C-Cdh-1 activity in ACC contributed to increase in GluR1 subunits of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors leading to up-regulation of synaptic activity. Furthermore, swollen myelinated fiber and collapsed axons were detected in neuronal cells in ACC, probably due to AMPA receptor trafficking-induced excitotoxicity. Micro-injection of Cdh-1 expressing recombinant lentivirus in ACC alleviated SNI-induced mechanical allodynia, normalized redistribution of GluR1 subunits of AMPA receptors and synaptic ultra-structure in ACC [134].
Hence, it may be insinuated that nerve injury decreases the expression of Cdh-1 in ACC, which in turn may increase the expression of AMPA receptor to induce neuropathic pain.
Go to :
DISCUSSION
There have been extensive studies in recent years to explore the efficacy of new targets in management of neuropathic pain (Table 1). These studies have been largely done in well established animal models that share clinical features of nerve injury such as CCI (correlated to carpal tunnel syndrome), spinal nerve injury, CFA-induced orofacial inflammatory pain (trigeminal neuralgia) and anticancer drugs-induced neuropathic pain. These models possess face validity, constructive validity and predictive validity. Based on these studies, it is possible that there may be activation of myriad of reactions in response to nerve injury that may interconnect to induce central sensitization. Although, a number of different targets have been projected as clinically potentially targets, yet the studies describing the interrelation among these targets are lacking. Most of the studies have targeted the preventive treatment before the development of neuropathic pain (i.e induction phase) [5263717484]. However, there have been very few studies documenting the influence of modulators of these targets in the maintenance phase of neuropathic pain i.e once the pain is already established. The study of Roh et al demonstrated the ineffectiveness of pharmacological modulator of sigma receptor antagonist in the maintenance phase of neuropathic pain [46]. Nevertheless, more elaborative studies are required to explore the role and understand mechanisms of these new targets in maintenance phase of neuropathic pain.
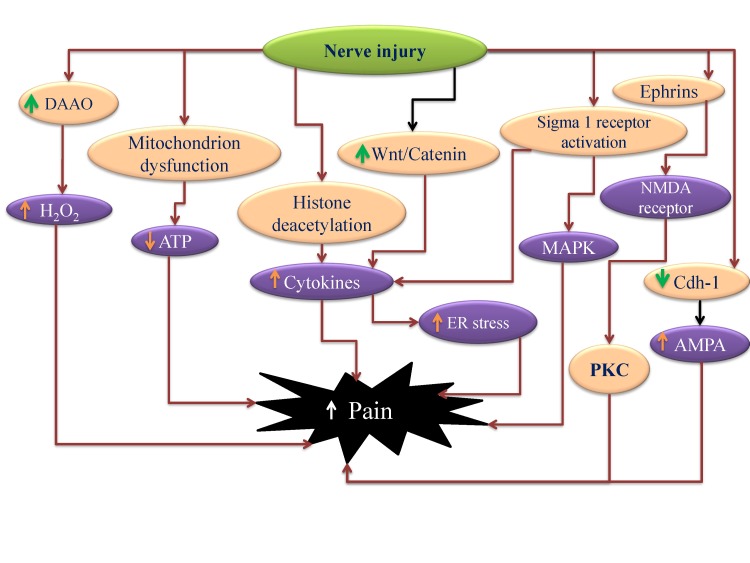
Research pertaining to these targets explores their individual roles in the disease, but close scrutiny of their mechanisms imply a significant correlation between these targets. It is possible that nerve injury may produce epigenetic changes such as histone deacetylation that may trigger the production of inflammatory mediators [34]. The activation of sigma receptors may also contribute in increasing cytokine production [62]. These increased levels of cytokines and free radicals, due to sigma receptor activation [63] and increased activity of DAAO [72135], may induce ER stress [83] which in turn may induce central sensitization. The development of neuroinflammation may also increase oxygen consumption and produce local hypoxia. This condition gets worsened due to mitochondrial dysfunction and decrease in Na+/K+ATPase activity [39]. Nerve injury may also affect genes or proteins affecting synaptic plasticity including decreased APC/C-Cdh1 activity in the ACC, which in turn may activate AMPA channel activity to increase pain perception [36]. The increased expression of ephrin B2 [72] along with β-catenin and menin [37] in the neurons of DRG and spinal cord may also modulate synaptic activity leading to increase in pain perception (Fig. 9). Moreover, membrane hyper-excitability caused by mitochondrial and bioenergetic dysfunction after nerve injury is a significant factor that may results in neuropathic pain.
Nevertheless, future studies are required to understand the precise interrelationship amongst these different targets. Furthermore, the comparative analysis of these targets may be done to understand the relative relevance of these targets in the pathophysiology of neuropathic pain. The pharmacological inhibitors of these therapeutic targets also exhibit adverse effects in patients such as histone deacetylase inhibitors produce nausea/vomiting, fatigue, and a transient decrease in platelet and white blood cell counts [136]; p38 inhibitors elevate liver enzymes, produce skin rashes and increase propensity to infections [137] and Wnt inhibitors possess teratogenic potential [138]. However, it is also worth mentioning that the most of inhibitors developed to inhibit these targets have been tested in preclinical studies and there is not enough studies delineating their possible side effects in clinical set up. Therefore, future studies may be designed to reveal the possible side effects of these pharmacological agents employed in neuropathic pain studies.
Go to :
 XML Download
XML Download