

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Atopic dermatitis (AD) is an inflammatory skin disease that affects up to 20% of children and 3% of adults (123). A hallmark of AD is dry and itchy skin with the disrupted skin barrier function. It is now considered that AD is a heterogeneous disease characterized by the activation of diverse cytokine signaling pathways, involving Th1, Th2, Th22, and Th17 cells, depending on the disease subtype (456). Notably, it has been reported that Asian patients with AD have much higher expression levels of Th22 and Th17 cell related cytokines in lesional skin, compared with those in American patients of the European origin (7). Additionally, new onset pediatric AD patients have robust mixed activation of Th2, Th22, and Th17 cells in AD skin lesions (89). Overall, these observations suggest that dysregulation of immune responses mediated by multi-polarized immune cells may participate in the diverse manifestations of AD.
IL-22 is a member of the IL-10 family of cytokines produced by Th17 and Th22 cells, innate lymphocytes that include γδ T cells and type 3 innate lymphoid cells (10111213141516). The IL-22 receptor (IL-22R) is expressed on epithelial cells, including keratinocytes, but not on immune cells, indicating an essential role for IL-22 signaling in mucosal barrier function (171819). IL-22 induces epithelial cell proliferation and expression of anti-apoptotic genes, and thus, promotes tissue repair activity and protects stem cells from injury (13202122). At the same time, it has been reported that the IL-22 signaling pathway participates in inflammatory skin diseases (23242526). For example, an intradermal injection of IL-22 in vivo causes keratinocyte proliferation and epidermal thickening (27). Administration of IL-22 in vitro also leads to keratinocyte proliferation and the thickening of human epidermis reconstituted in a three-dimensional matrix (2829), similar to the changes in the lesional skin of AD and psoriasis patients. Indeed, Il22 mRNA expression and T cells that produce IL-22 are significantly increased in skin lesions of patients with AD (303132). Additionally, serum IL-22 levels are also elevated in patients with AD (3334).
In line with these reports, we will discuss our recent findings on the contribution of IL-22/Th22 cells to allergic skin inflammation as well as Th22 cell polarization obtained in the murine model of AD induced by epicutaneous sensitization of allergen (35). We will present evidence that targeting of IL-22 signaling can be a plausible therapeutic option for AD. Finally, we will finish this review summarizing major clinical approaches currently being developed for treating AD.
THE SKIN AS A MAJOR ROUTE OF ALLERGEN SENSITIZATION FOR ALLERGIC DISEASES
The skin is considered as the largest immunological organ that acts as a barrier between the body and external environments, protecting against chemical and physical insults as well as against pathogenic microbes (3637). Defects in skin barrier function and abnormalities of skin immune systems give rise to skin inflammation and microbial infection (3839). AD generally tends to precede other allergic diseases such as asthma, allergic rhinitis and others, known as the atopic march (40414243), implying that the skin may be a crucial priming site in AD as well as in allergies of different parts of the body. In fact, most infants and children with AD history have a higher tendency to develop asthma, allergic rhinitis (4044) and other allergic conditions than infants without AD. Moreover, we and other groups experimentally proved that mechanical skin injury with tape stripping, a surrogate of scratching in AD, exacerbates the defect in skin barrier function, leads to the penetration of the allergen through the damaged skin, and induces the release of a range of cytokines and chemokines that drive immune responses to cutaneously immunized antigens (35454647). This AD-like skin inflammation subsequently stimulates allergic manifestations such as airway hyper-responsiveness (484950) and food allergy (5152). Overall, accumulated clinical and experimental findings unequivocally support the notion that the skin is a major sensitization site for allergic diseases that affect different parts of the body.
MOUSE MODEL OF AD TRIGGERED BY EPICUTANEOUS SENSITIZATION
Our understanding of human diseases has been enormously expanded by in-depth studies in animal models that are invaluable tools for unveiling pathogenic mechanisms and finding potential treatment targets. Therefore, to find potent target(s) and develop medicines for treating AD, it is essential to elucidate AD pathogenesis by establishing appropriate animal models that faithfully recapitulate the hallmark features of AD. To satisfy this demand, Spergel and colleagues (53) have developed a mouse model of AD induced by repeated epicutaneous sensitization of the injured skin inflicted by tape-stripping with ovalbumin (OVA). This model displays many features of human AD described below. Mechanical injury of the skin with tape stripping, a surrogate of scratching in patients with AD, enhanced the release of various pro-inflammatory cytokines and chemokines, which are regarded as important initiating factors for allergen-specific immune responses and allergic skin inflammation. Mice epicutaneously sensitized with allergen developed increased scratching behavior and skin lesions that exhibited many cardinal features of AD, including increased epidermal and dermal thickness, infiltration of a mixture of polarized CD4+ T cells and eosinophils as well as the expression of Th2 (53), Th17 (48), and Th22 (3550) cell derived cytokines with minimal or no change in IFN-γ level (53). Systemically, serum OVA-specific IgG1, IgE, and IgG2a were elevated in this model (53), as typically described in AD patients. Furthermore, OVA-sensitized mice developed higher airway hyper-responsiveness following a challenge with OVA (485053), frequently observed in asthmatic patients with AD history. Additionally, epicutaneous sensitization with allergen enhanced IgE-mediated mast cell degranulation and promoted mast cell-dependent anaphylaxis elicited by oral challenge (5152), which was similar to the food-induced anaphylaxis in patients with AD. Thus, this model has histological, immunological, and clinical features of human AD and can be utilized to gain better insights into the mechanisms of AD pathogenesis and allergic diseases associated with AD. Furthermore, this model can be utilized to find therapeutic targets and to develop medicines for the treatment of AD and allergic diseases associated with it.
POTENTIAL ROLE OF IL-22 AND TH22 CELLS IN AD
IL-22 is a member of the IL-10 family of cytokines, which was initially thought to be produced by Th1 cells (54). However, it was found that this cytokine is secreted in high quantities by Th17 cells (55). Interestingly, IL-22 and IL-17 are not always co-expressed and their expression are regulated differently in humans (5657). Furthermore, identification of Th22 cells producing IL-22 without the concomitant production of IFN-γ, IL-4, or IL-17 has attracted much attention due to their potential involvement in skin homeostasis and pathogenesis of AD (1516). Whereas the retinoid-related orphan receptor C is the master transcriptional factor for Th17 cell differentiation, the aryl-hydrocarbon receptor (AHR) is a critical factor for Th22 cell polarization in humans (16). Indeed, activation of the AHR induces a robust IL-22 expression, whereas it inhibits IL-17 production (16). Furthermore, it was reported that a subset of memory CD4+ T cells producing IL-22 express skin homing chemokine receptors C-C chemokine receptor (CCR) 10, CCR6, and CCR4 that promote their infiltration to skin (1516). Thus, it implies that Th22 cells may have a potential role in skin homeostasis and inflammation. Interestingly, the IL-22 receptor is expressed on epithelial cells, including keratinocytes in the skin, but not in immune cells (19), implying a potential role of IL-22 signaling in skin barrier functions contributed by keratinocytes. Th22 cells and IL-22-producing CD8+ cells are the main source of IL-22 in AD (31). Additionally, a high percentage of circulating Th22 cells and IL-22-producing CD8+ T cells from patients with AD co-express IL-13 (58). Moreover, the number of Th22 cells and IL-22+CD8+ T-cell positively correlates with AD disease severity (31), and IL-22, highly expressed in the affected skin of patients suffering from AD, is deeply involved in epidermal hyperplasia and barrier defects (5759). More importantly, therapeutic success at the early stage of the clinical trial of ILV-094, an anti-IL-22 antibody, demonstrated that IL-22 indeed contributes to the pathogenesis and symptoms of AD (6061). We also demonstrated that epicutaneous sensitization by the application of allergen to mechanically injured mouse skin resulted in elevated serum IL-22 levels and accumulation of CD3+CD4+IL-22+ T cells in allergen-sensitized skin, which is essential for epidermal thickening and keratinocyte proliferation, as both were absent in Il22−/− mice (35). In complementary experiments, we also showed that keratinocyte proliferation and epidermal thickening were similarly induced in OVA-challenged skin of naïve recipients of Th22 cells polarized in vitro (35). Furthermore, our recent findings demonstrated that epicutaneous immunization induced polarization of IL-22-producing T cells that subsequently contributed to airway hyper-responsiveness in allergen-challenged mice (50). These data support the view that targeting IL-22 in AD may be promising for the treatment of AD and other allergic diseases, including asthma.
PLAUSIBLE MECHANISMS OF TH22 CELL POLARIZATION IN THE SKIN
Since the discovery of the novel Th22 cell subset in human skin, the knowledge of IL-22/Th22 cell biology has increased considerably. A recent report demonstrated that human Langerhans cells (LCs) efficiently induce the expansion of memory Th22 cells and stimulate the polarization of Th22 cells from naive CD4+ T cells (62). Furthermore, recent findings showed that tissue-resident CD5-expressing dendritic cells (DCs), a subtype of the healthy human LCs and dermal skin cells can induce Th22 cells and cytotoxic T cells (63). In addition, it has been shown that the expression of CD1a, a lipid-presenting molecule, in a subtype of LCs is critical for the expansion of IL-22-producing T cells (64). However, the mechanism of differentiation of antigen-specific IL-22-producing T cells was not determined in that study. To examine in detail the mechanisms responsible for Th22 cell polarization, we utilized a mouse model of AD elicited by the application of antigen to mechanically injured skin. In this model, we have demonstrated a sequence of mechanisms leading to IL-22-dependent epidermal thickening and polarization of allergen-specific Th22 cells. Endogenous TLR4 ligands released by mechanical skin injury, a surrogate of scratching in AD, cause keratinocytes to release IL-23 that drives allergen-captured migratory skin DCs to produce endogenous IL-23 and polarize naïve CD4+ T cells to antigen specific CD4+ T cells. The latter cells, in turn, mount IL-22 response to cutaneously introduced antigen, causing keratinocyte proliferation and epidermal thickening, a hallmark feature of AD (35). We also observed that the IL-23 receptor (IL-23R) is expressed on a subpopulation of DCs in the skin and skin draining lymph nodes (DLNs) in mouse, but is not detectable on splenic DCs (35). We further proved that recombinant IL-23, as well as IL-23 released from explants of mechanically injured skin, directly polarizes DCs from skin DLNs in vitro to drive IL-22 production by naïve CD4+ T cells (35). More importantly, the relevance of this pathway to the pathophysiology of AD in humans is supported by the observation that human keratinocytes express biologically active IL-23 and our findings that a fraction of epidermal LCs and dermal DCs in normal human skin express IL-23R on their surface, as exogenous IL-23 polarizes LCs and drives IL-22 production by naïve CD4+ T cells (35). Overall, our data suggest that IL-23 released by keratinocytes following mechanical injury primes a subtype of IL-23R+ skin-derived migratory DCs to express endogenous IL-23, which polarizes them and initiates an IL-22 response to introduced antigen via damaged skin (35). The priming action of keratinocyte-derived IL-23 on skin DCs that induces an IL-22 immune response (35) parallels the priming action of keratinocyte-derived thymic stromal lymphopoietin (6566) and IL-33 (67) on DCs that stimulates a Th2 immune response. Thus, the release of cytokines by keratinocytes is a key early event in the development of adaptive immune response to cutaneous sensitization with allergen.
POTENTIAL THERAPEUTIC TARGETING OF IL-22/TH22 SIGNALING PATHWAYS IN AD
As mentioned above, IL-22 is highly expressed in the severely affected skin of individuals suffering from AD (60). Chronic AD is characterized by the conversion of the immune response from Th2 cell-dominated to mixed activation of Th1, Th22, and Th2 cells in the affected skin (3132). We also proved that IL-22 is a major player in epidermal acanthosis observed in the sensitized skin of mice (35) (Fig. 1). In line with these observations, a randomized, double-blind, placebo-controlled clinical study of an anti-IL-22 mAb (ILV-094) showed a substantial improvement in patients severely affected with AD (61). Thus, it is likely that a subset of patients with AD with more polarization toward Th22 cell cytokine pathways might be responsive to the direct blockade of IL-22 signaling pathway (6061). Other potential therapeutic approaches to the IL-22 mediated skin inflammation may include targeting of IL-23, a key cytokine in the process of Th22 cell polarization in the sensitized skin as described above (35) (Fig. 1). Furthermore, blockade of CC-chemokine ligand 20 and its cognate receptor CCR6 that attracts IL-22-producing cells to the sensitized skin can be another therapeutic option for AD (68) (Fig. 1). Finally, because the AHR is a key transcription factor for both Th22 polarization and IL-22 production (1516), its antagonists, including CH223191 (69), can be utilized to suppress Th22 cell polarization or to block IL-22 production for the treatment of AD that involves Th22 cells and IL-22 (Fig. 1).
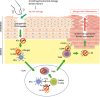 | Figure 1Potential therapeutic targeting of Th22/IL-22 signaling pathways in AD. Skin barrier defects caused by scratching or genetic mutations lead to penetration of external antigens and keratinocyte production of IL-23 via endogenous TLR4 ligand/TLR4 axis. A subset of IL-23R expressing DCs are activated and triggers AHR dependent Th22 immune response. Skin infiltrated CCR6+ Th22 cells induce epidermal hyperplasia and barrier dysfunction via IL-22/IL-22R signaling axis. Targeting TLR4/IL-23/Th22/IL-22 as well as CCL20/CCR6 pathways might be a promising strategy to overcome atopic skin inflammation.
|
ALTERNATIVE THERAPEUTIC APPROACHES FOR THE TREATMENT OF AD
Our increasing understanding of the pathogenic mechanisms of AD expanded the pipeline of new and more targeted therapies (70717273). Targeting therapies based on various mechanisms of action are being developed to suppress immune responses in AD (7071). Dupilumab, the first effective biologic targeting the IL-4/IL-13 receptor was approved by Food and Drug Administration for the treatment of moderate-to-severe AD (74). In a randomized, double-blind, placebo-controlled phase II study of nemolizumab targeting IL-31 signaling involved in itching, adult patients with moderate-to-severe AD showed the improvement of pruritus (7576). In addition, it was known that the JAK/STAT signaling axis is deeply involved in the dysregulation of immune responses in AD (7778). Based on this information, several therapeutic agents targeting TYK2, JAK1, JAK2, and JAK3 are being evaluated for treating moderate-to-severe AD (707273). The histamine/histamine H4 receptor (H4R) signaling has been linked to pruritus and inflammation in several preclinical AD murine models (798081). Thus, it can be a promising target for treating AD. In line with this assumption, oral administration of the H4R antagonist ZPL-389 significantly reduced pruritus in AD patients during clinical trials (82). Another type of H4R antagonist, JW1601, exhibited strong anti-pruritic and anti-inflammatory efficacies in several preclinical mouse studies (personal communication and unpublished data) and its phase I clinical trial will be shortly underway after the completion of good laboratory practice preclinical toxicity studies.
FUTURE PERSPECTIVES
In this review, we considered current available clinical and preclinical data supporting the notion that IL-22 may play a key role in skin inflammation by regulating epidermal thickness and skin barrier function. Thus, IL-22 blockade may prove beneficial to patients with AD. In particular, a recent clinical trial showed that treatment with fezakinumab, a monoclonal antibody against IL-22, consistently improved clinical disease scores in adults with moderate-to-severe AD (6061). Despite this benefit, it may be limited to the subtype of AD patients that are affected by dysregulated IL-22/Th22 cell immune response (60). Additionally, because individual patients experience AD symptoms of heterogeneous severity, exhibit different patterns of immune cell activation, and are influenced by distinct genetic risk factors, it is important to advance our knowledge of AD pathogenesis by using appropriate animal models and carefully designed clinical studies. Precise sub-phenotyping and determination of biomarkers in individual patients based on immunophenotypes and genomic patterns should be considered for further development and application of precision medicine in the management of AD.
 XML Download
XML Download