

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Needs for better treatment options for children
ICH E11 states that “Pediatric patients should be given medicines that have been appropriately evaluated for their use. Safe and effective pharmacotherapy in pediatric patients requires the timely development of information on the proper use of medicinal products in pediatric patients of various ages and, often, the development of pediatric formulations of those products. … …. this should be done without compromising the well-being of pediatric patients participating in clinical studies. This responsibility is shared by companies, regulatory authorities, health professionals, and society as a whole.”[1]
In US, through continuing effort by the government as well as other stakeholders, 764 new pediatric labeling changes have been made as of Oct 31, 2018, among which 697 with new pediatric studies and 67 with no new pediatric studies.[2] However, there are many more drugs that have been used for a substantial amount of time as off-label uses, for which the patents have expired, and numerous copy drugs are being sold. These drugs are used as off-label, without adequate information regarding their efficacy or safety, especially in children. There exists public, regulatory and medical providers'[3] concern for safety, as well as for inadequate efficacy. It is imperative to generate evidence for safety and efficacy for these off-label drugs.
On the other hand, for pharmaceutical companies, pediatric development itself is difficult, adds financial burden, but with less return on investment in general. It also encompasses the need for development of new formulations for children, which is another burden to the companies. Perhaps not getting indication expansion for pediatric uses or even in adults would not incur any damage to the finance of the companies, since as long as the companies do not unlawfully promote off-label uses, physicians will prescribe the drugs off-label, and sales will be maintained anyway.[4]
Conducting clinical trials is difficult and may not always be feasible
In order to make label changes for indication expansion, generation of evidence of efficacy and safety through well-designed randomized controlled trials is considered ideal for acceptance by regulatory agencies. However, conducting clinical trials for children is challenging, at least operationally. There are difficulties conducting trials in children due to the limitations such as a small population size usually not quite enough to obtain robust and interpretable results, developmental and maturational changes, phenotypic variability, poorly understood natural history, lack of appropriate biomarkers, outcome measures and endpoints, difficulties in defining control groups, ethical concerns with use of placebo, healthy children, confirming child's willingness and getting informed consents, sample collection issues, etc.[5]
Alternative approaches are to be sought.
ICH E11R1 with addendum describes the use of existing knowledge in pediatric drug development; that is, the use of extrapolation and modelling & simulation (M&S).[1] “Pediatric extrapolation” is defined as an approach to providing evidence in support of effective and safe use of drugs in the pediatric population when the course of the disease and the expected response to a drug would be sufficiently similar in the pediatric and reference population. In support of the use of pediatric extrapolation, there are several important questions to be asked to identify whether additional supportive data are needed. We should consider and acquire evidence for a common pathophysiology of disease, natural history, and similarity of the disease course; strength of the evidence of efficacy & biomarker or surrogate endpoint in the reference populations; evidence for a similar exposure-response; uncertainties and/or limitations of the existing data (e.g., clinical or historical data and published literature); additional information to be generated for the acceptability of the extrapolation approach. M&S can be used to quantify available information and to design pediatric clinical studies and/or the dosing strategy. It can help avoid unnecessary pediatric studies and help ensure generation of appropriate data from small number of patients. Well conducted M&S can inform on the pharmacokinetics, pharmacodynamics, efficacy and safety of a drug.
Extrapolation strategy introduced to optimize pediatric drug development
Similar concept as described in E11R1 has been introduced as early as in 1992 in the US. The proposal of Federal Register of October 16, 1992 (57 FR 47423) and the final regulation in 1994 (59 FR 238) allowed pediatric claims based not only on adequate and well-controlled studies in the pediatric population but also on such trials in adults, provided that the course of the disease and the drug's effects are sufficiently similar in the pediatric and adult populations to permit extrapolation from the adult efficacy data to pediatric patients. Pharmacokinetic data may be used for determination of an appropriate pediatric dosage, and additional pediatric safety information must also be submitted.
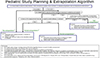
There are three possible approaches described to the use of extrapolation of efficacy from adult to the pediatric population as shown in the diagram. Briefly, if no extrapolation can be used, two adequate well-controlled efficacy and safety trials in children plus pharmacokinetic data are needed. For partial extrapolation, a single adequate, well-controlled efficacy and safety trial plus pharmacokinetic data or demonstration of exposure/response in defined situations. And for complete extrapolation, pharmacokinetic and safety data are required (Fig. 1).[6]
The use of data sources other than randomized controlled trials (RCT)
Dr. Frieden illustrates limitations of RCTs and suggests the use of alternative data sources for health decision making.[7] In a sense, regulatory approval for new drugs falls into the same category when it comes to making health-related decisions. We have been trained over the years and blindly attached to the RCTs as the best and only reliable evidence when it comes to drug approval reviews. However, realizing the weaknesses of RCTs in the real world such as limited validity in subject population and conditions, resource-intensiveness with regard to cost and time required, and impracticality in certain conditions like rare diseases and in some cases pediatric populations, it is about time to open our eyes and seek for other sources to rely on and make them better to suit our purposes.
At the 2016 American College of Clinical Pharmacology Annual Meeting Symposium, there was a discussion on the utility of big data or real-world data for informing clinical trial design and substitute for efficacy and safety data typically obtained in clinical trials, including future pediatric drug development. Some of the promising areas of the application of big data were generation of hypotheses, informing inclusion/exclusion criteria, duration of study, and selection of appropriate control; support for the use of pediatric extrapolation; development of non-concurrent control groups (e.g., historical or external control) for pediatric clinical trials; and supportive short-term and long-term safety data.[5]
The 21st century cares act and expanding indications using real world data (RWD)/real world evidence (RWE)
In 2016, US decrees the 21st Century Cures Act. And in 21 USC 355g: Utilizing real world evidence (Section 3022) commits the FDA to evaluate the potential use of RWE to help to support approval of a new indication for a drug approved under section 355(c), which is 21 USC 355c: Research into pediatric uses for drugs and biological products, and to help to support or satisfy postapproval study requirements. This act triggers a series of activities that will shape a new paradigm for the future drug regulatory approval.
Use of RWD/RWE offers much broader and diverse patient experiences compared with traditional phase 3 RCTs. In addition, the big data source used for RWD/RWE provides vary large sample sizes that potentially enable detection of infrequent events as well as drug interactions. And unlike RCTs it requires low resource intensity as in observational DB studies, where data from routine interactions of patients with their health care system can be utilized, or in pragmatic clinical trials, where there is no blinding and data from patients' usual health care such as EHR and claims can be utilized. RWE from Sentinel, which provides access to large claims database to assess potential safety signals, might be used for evaluation of safety of marketed products. Prospectively collected registry data or external control DB using observational clinical dataset could be used to define natural history or to provide historical control for interventional single arm trials in oncology or rare diseases studies. Avelumab (a programmed death ligand [PD-L1] blocking human IgG1 lambda monoclonal antibody) as monotherapy for metastatic Merkel cell carcinoma (mMCC) received accelerated approval in the US and Europe. For the JAVELIN Merkel 200 trial, RW data of mMCC patients that received chemotherapy were used to show survival benefit of the immunotherapy over chemotherapy and were offered to regulators as external control, not a formal comparator arm.[8] Label expansion in paroxysmal nocturnal hemoglobinuria, a rare and life-threatening disease, was granted by the EMA for eculizumab based on the data from an observational, international disease registry.[9]
However, there are numerous issues to consider for using non-interventional observational studies to support regulatory decisions, such as selecting appropriate research questions, finding adequate and available DB suitable for analysis of endpoints, selecting appropriate patient populations with comparable medical care, adequate duration for follow-up and others, not to mention data accuracy, completeness and robustness, and research integrity.
Randomized pragmatic trials
Califf and Sugarman defines a pragmatic clinical trial (PCT) as “designed for the primary purpose of informing decision-makers regarding the comparative balance of benefits, burdens and risks of a biomedical or behavioral health intervention at the individual or population level”.[10] One of the examples of pragmatic clinical trial is ADAPTABLE (Aspirin Dosing: A Patient-centric Trial Assessing Benefits and Long-Term Effectiveness). It compares the effectiveness of two different daily doses of aspirin (81 mg and 325 mg) in patients with atherosclerotic cardiovascular disease (ASCVD). The trial utilizes EHR data that have been standardized according to a common format. In the course of the trial, routine clinical practice is not to be interfered with. Patients who meet criteria for trial enrollment are identified using search algorithms. Patients are randomized 1:1 to either dose and not blinded. Patients are followed up for patient reported outcomes every 3 or 6 months, supplemented with searches of EHR, CDM and claims data. Over 3 to 4 years of follow up composite of all-cause mortality, hospitalization for MI or for stroke will be measure for primary endpoint, while hospitalization for major bleeding as primary safety endpoint.
Major differences between PCT and RCT, as mentioned by Dreyer et al, would be that PCT does not use placebos and that surrogate markers are rarely used. In addition, PCT has stronger external validity by including more diverse patients, medical care providers, and settings. Site and data monitoring is rarely performed or remotely conducted (SDV around 20%). Open label treatments and less SDV drive cost down. For outcomes of interest such as major adverse cardiac events that are generally evident even in real world settings, PCT is likely to generate evidence reliable enough for regulatory decision.[11]
Randomized registry trial
The registry-based randomized trial, or randomized registry trial (RRT), aims to approach the statistical rigor of randomization, whereas expediting and enhancing patient enrollment, minimizing cost, and addressing the generalizability of findings.[1213] By including a randomization module in a large inclusive clinical registry with unselected consecutive enrolment, the advantages of a prospective randomized trial can be combined with the strengths of a large-scale all-comers clinical registry.[14] Thrombus Aspiration in ST-Elevation Myocardial Infarction in Scandinavia (TASTE) trial utilized their already existing Swedish angiography and angioplasty registry (SCAAR) platform for blind evaluation of end points as well as for randomization of treatments to STEMI patients, either to conventional PCI or to thrombus aspiration + PCI. The primary endpoint is time to all-cause death at 30 days.[15]
Solutions for Korea
In Korea, the way that the regulatory authority demonstrates its care for children, unfortunately, is by prohibiting off-label uses, by branding them as contraindications based on the mere fact that safety and efficacy have not been established in the age groups. Unlike other countries, where once a drug is approved, off-label uses are regarded as a part of the practice of medicine and not under regulatory authority but at the discretion of physicians for medical necessity or for possible expansion of indications, in Korea regulatory authority takes the responsibility of protecting children by blocking potentially inappropriate, but also potentially appropriate and necessary, off-label uses from physicians' indiscretion at its source, not quite weighing out whether their act of prohibiting off-label use at all can be justified over depriving children of any chances of appropriate treatment options.
In attempts to minimize off-label uses, and at the same time, to straighten out per se the situation in which uncertainty prevails whether to allow or not the current off-label uses, the government has been encouraging conduct of clinical trials to generate sufficient evidence for making label changes either to approve expanded indications or disapprove off-label uses at all. However, conducting well-controlled clinical studies in children and getting the evidence for safety and evidence at the level of quality, quantity, science and rationale that satisfy regulatory approval standards, and at the same time not incriminating on ethical issues, is not easy, if not almost impossible.
Korea has only a handful of pharmaceutical companies that develop new drugs. Making the laws that give incentives to pharmaceutical companies that conduct trials for pediatric indications as well as require pediatric studies will help, but only to a certain and small extent, to solve the issues of off-label uses in children overall. Most of the drugs used off-label are imports of overseas companies or copy drugs that are already off-patent. It is unlikely that any incentive or requirements made locally will have any impact on the foreign companies to initiate pediatric development. Pharmaceutical companies are not interested in investing money into label changes or indication expansion for a country like Korea even if the same changes or expansion have been implemented in the original label. The reason being the financial burden to carry out additional postmarketing surveillance studies.
We need a change in the paradigm of regulatory approval of indication expansion.
As mentioned previously in this article, the regulatory agency needs to accept the situation where evidence from studies other than RCT can be utilized to satisfy the regulatory approval standards for safety and efficacy. As the 21st Century Care Act in the US acknowledges the use of real world evidence should be incorporated for pediatric drug development and indication expansion, Korea has to embrace and join the global movement for the safer and better drugs for children. Regulatory eyes should not be fixed only to the past, equally incomplete and limited tools as RCTs. More diverse sources and methodologies with the help of evolving technologies should be adopted to generate evidence that supplements or replaces the evidence obtained through classical RCTs. Off-label use registries should be upgraded, modified and redesigned to fit for purposes of getting evidence for safety, and in certain cases for efficacy. Pragmatic trials should be more widely employed to generate relevant evidence. For that matter, alignment of reimbursement data with EHR/EMR as well as standardization and sharing of medical records of individual hospitals for research purposes should be a prerequisite.
 XML Download
XML Download