

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Chronic lymphocytic leukemia (CLL) is the most common leukemia among adults [1], and is characterized by the clonal proliferation of mature B lymphocytes [2]. This disease typically affects elderly patients, with a dramatic increase in its incidence at the age of >60 years, and the male:female ratio in patients is 1.5–2:1. The clinical features of CLL are lymphadenopathy, splenomegaly, fatigue, infection, and anemia [3]. However, 25–50% of patients are asymptomatic and the diagnosis is frequently made via routine blood testing [4]. Furthermore, CLL has a highly variable clinical course that ranges from an indolent disease requiring no treatment to a rapidly progressive disease that requires early intervention [5].
According to the International Workshop on CLL, a diagnosis requires the clonal expansion (≥5×109/L) of B lymphocytes in the peripheral blood that express the B-cell antigens (CD19, CD20dim, CD23) plus CD5 and weak expression of surface immunoglobulin (IgM or IgD) [6]. The Rai and Binet staging systems are the main clinical systems for predicting the prognosis of patients with CLL, and these systems are based on a physical examination and blood test results [7]. Patients can also be stratified into high-risk or low-risk groups based on other markers, such as B2 microglobulin, CD38, and ZAP70. Cytogenetic testing also plays an important role in defining distinct subsets of CLL [8], based on cytogenetic abnormalities that can be detected using conventional metaphase karyotyping. However, interphase fluorescent in situ hybridization (FISH) reportedly has a much higher sensitivity for detecting many of these abnormalities, especially deletion of 17p (del 17p) [3].
Patients with del 17p have a poor prognosis, as this deletion at 17p13 leads to loss of p53 function in CLL, which contributes to disease progression, poor treatment response, and shortened survival [9]. The prevalence of del 17p in CLL has been estimated to be 16.6% [3], and this change is associated with high-risk disease that does not respond well to standard conventional regimens [10]. However, most studies regarding CLL with del 17p have examined Western populations, despite the fact that there are noticeable differences in disease biology, clinical presentation, disease progression, and outcomes in various geographical population groups. Therefore, the present study aimed to evaluate the clinicohematological features of patients with CLL with or without del 17p in Pakistan, in order to identify factors that could predict their treatment response, disease progression, and overall survival. Because there is a lack of data regarding CLL with del 17p in Pakistan, this information may help improve local treatment protocols and patient prognoses.
MATERIALS AND METHODS
Patients
This prospective study evaluated 130 patients who were treated between January 2013 and December 2015. The patients belonged to various ethnic groups and were 45–80 years old. All patients had newly diagnosed CLL according to the International Workshop on Chronic Lymphocytic Leukaemia IWCLL criteria, based on persistent lymphocytosis, lymphocyte morphology on peripheral blood smears, and immunophenotyping results. None of the patients had a history of previous treatment. All patients received a detailed explanation of the study and provided written informed consent for the data collection and testing. This study's protocol was approved by the Ethical Review Committee of the Armed Forces Institute of Pathology (Rawalpindi, Pakistan).
Clinicohematological parameters
A detailed history was collected and a physical examination was performed for each patient, and their symptoms and signs were noted. The diagnostic work-up included a complete blood count, peripheral blood smear, chest radiography, and abdominal ultrasonography. Serum biochemical testing was performed for all patients. In addition, a bone marrow examination was performed, as well as testing for B2 microglobulin, CD38, and ZAP70.
Cytogenetics and FISH
Cytogenetic analysis was performed using the conventional methods. Interphase FISH testing was performed using blood or bone marrow specimens that were processed via the standard methods for cultured samples. The FISH assay was performed using 10 µL of the Meta systems XL p53 probe, and a total of 500 nuclei per assay were analyzed using a fluorescent microscope with an orange-green spectrum filter. Patients with failed culture results were excluded from the study. The del 17p clone size was noted for each case.
Treatment and monitoring
The indications for treatment included an increase in the white blood cell count with a <6-month lymphocyte doubling time; development of B symptoms; symptomatic anemia and/or thrombocytopenia; or progressive enlargement of lymph nodes, the spleen, and/or the liver. The time from the diagnosis to the first treatment was recorded, with the therapies including chlorambucil, fludarabine plus cyclophosphamide plus rituximab, bendamustine plus rituximab, and glucocorticoids. Overall response was defined as a ≥50% reduction in the peripheral absolute lymphocyte count or the size of previously enlarged nodes/spleen/liver, relative to the pre-treatment values.
Statistical analysis
The statistical analyses were performed using IBM SPSS software (Statistical Package for Social Sciences, version 20, IBM, Armonk, NY, USA). The patients' baseline characteristics and prognostic factors were compared using the chi-square test. All p-values were 2-sided and differences were considered statistically significant at P<0.05. The event-free survival (EFS) interval was calculated from the start of treatment to the first instance of disease progression or complication. Kaplan-Meier curves were plotted for the EFS intervals and compared for the potential prognostic variables using the log-rank test. The 2-year overall survival (OS) rate was calculated for the 2 years after the date of diagnosis.
RESULTS
The study included 130 patients with a median age of 65 years. Most patients were male (104 patients, 80%) and the remaining 26 patients were female (20%). The mean absolute lymphocyte count was 89.12±28.06 (×109/L), the mean hemoglobin level was 10.31±1.48 g/dL, and the mean platelet count was 154±36.89 (×109/L).
Interphase FISH analysis was performed for all patients, which detected del 17p in 24 patients (18.5%). In addition, the FISH analysis identified del 13q in 3 patients and del 11q in 2 patients. One patient had a complex karyotype based on conventional cytogenetic analysis. The clone size analyses revealed that 33% of the patients had <25% 17p– cells and 67% of the patients had >25% 17p– cells.
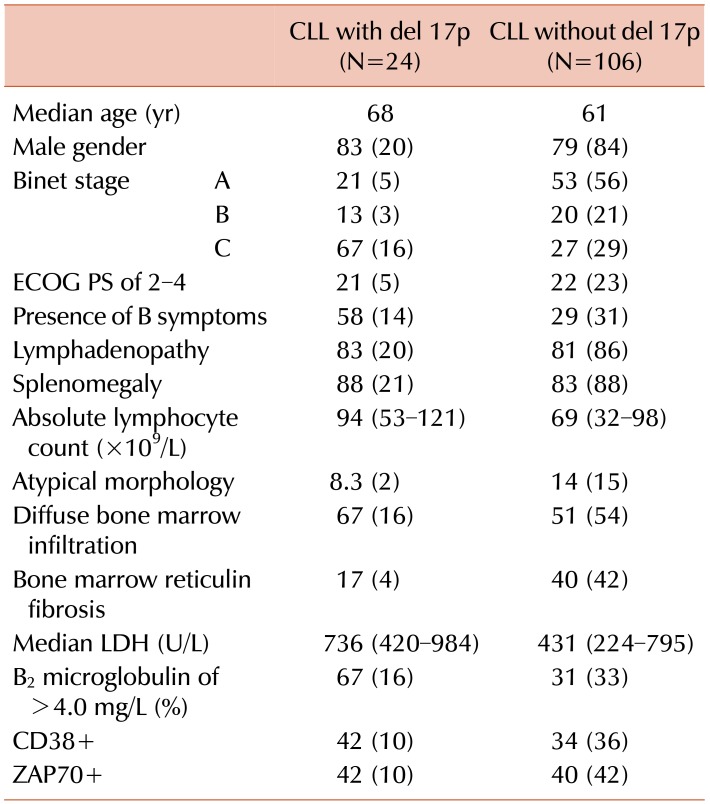
Table 1 shows the clinicohematological parameters of the patients with CLL with or without del 17p. A comparison of these groups revealed that del 17p was significantly associated with age >65 years (P=0.01), advanced clinical stage (P=0.0001), presence of B symptoms (P=0.007), and elevated B2 microglobulin levels (P=0.001). However, del 17p was not significantly associated with gender (P=0.65), CD38 positivity (P=0.47), or ZAP70 positivity (P=0.85).
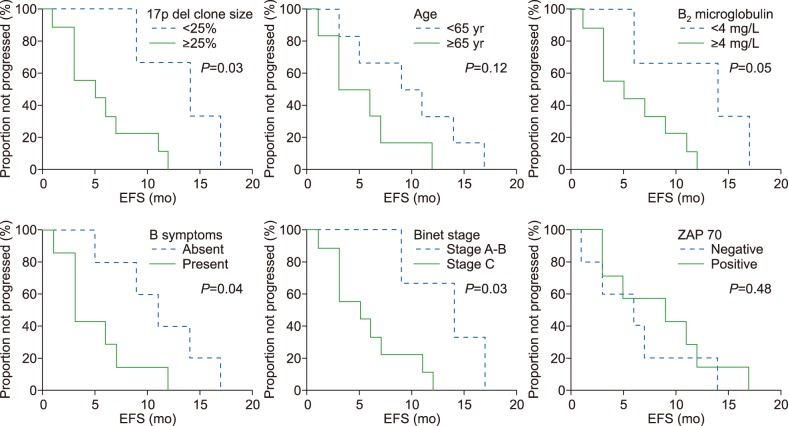
Among the 24 patients with CLL and del 17p, 4 patients were lost to follow-up. During a 24-month follow-up period, 14 patients (70%) underwent treatment and 6 patients (30%) did not require treatment. The median time from diagnosis to the start of treatment was 11 months (range, 0–24 mo). The overall response rate was 64.3% (N=9), although 5 patients (35.7%) were refractory to primary therapy. The 2-year EFS rate was 11%, with a median interval of 9 months (range, 1–24 mo). Fig. 1 shows the study variables and their associations with EFS. Seven patients died during the 24-month follow-up period. The causes of death included infections (pneumonia and tuberculosis), disease progression, and Richter's transformation. The 2-year OS rate was 65%. Univariable analysis revealed that poor OS was significantly associated with B symptoms (P=0.04), although OS was not significantly associated with age, gender, Binet stage, del 17p clone size, CD38 status, or ZAP70 status.
DISCUSSION
During the last decade, an improved understanding of CLL biology has led to the identification of genetic and molecular factors that predict the prognosis of CLL patients [11]. However, conventional metaphase cytogenetic analyses underestimate chromosomal aberrations in CLL patients, as the detection of abnormalities is limited by the low mitotic activity of CLL cells, whereas FISH analysis using interphase cells can identify up to 80% of chromosomal abnormalities [3]. Thus, FISH is considered much more sensitive for CLL, with better ability to detect genomic aberrations, which makes it the preferred method for reliably identifying chromosomal aberrations in CLL [12]. Among the various cytogenetic abnormalities that can be detected in CLL, del 17p is thought to be associated with the poorest prognosis [13].
Recent advances in treatment protocols, and the introduction of fludarabine/cyclophosphamide/rituximab as first-line therapy, have led to better rates of complete remission and OS among patients with CLL [14]. However, patients with del 17p are challenging to treat, as they do not respond well to conventional treatment and experience poor progression-free and OS outcomes [15]. Therefore, these patients require alternate treatments, with targeted therapy and novel agents potentially producing better outcomes [16]. Thus, early detection of del 17p can help identify high-risk cases that should undergo appropriate treatment to improve their survival [17].
To the best of our knowledge, there are no available FISH-based data regarding the frequency of del 17p among patients with CLL in Pakistan, or data regarding the clinicohematological characteristics and outcomes for this high-risk group of patients. In this context, the Armed Forces Institute of Pathology is a large institution in a northern region of Pakistan, which treats a large number of patients from throughout the country who have various ethnic backgrounds. Therefore, we evaluated the frequency of del 17p among patients who were treated at our center, and summarized their clinicohematological characteristics and clinical outcomes.
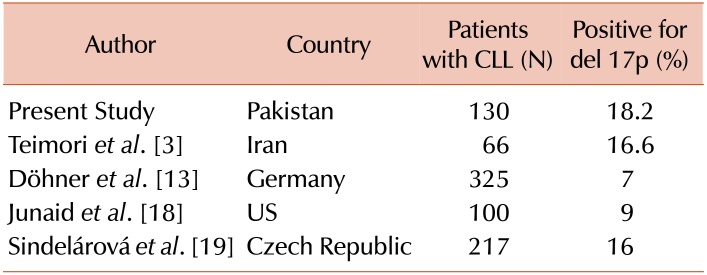
The present study included 130 newly diagnosed cases of CLL, with 24 patients (18.5%) being positive for del 17p. These results agree with the findings of a relatively local study by Teimori et al. [3], who reported a frequency of 16.6% in an Iranian population. However, Table 2 shows that lower frequencies of del 17p have been reported by Döhner et al. [13] in Germany (7%) and by Junaid et al. [18] in the US (9%). Nevertheless, Sindelárová et al. [19] have reported a frequency of 16% in the Czech Republic, which agrees with our findings.
The present study revealed a patient age range of 45–75 years, with a mean age of 64.18 years. Delgado et al. [20] have reported a mean patient age of 68 years, which is similar to our findings. Furthermore, Rozman and Montserrat [21] reported a mean patient age of 65 years, and Teimori et al. [3] reported a mean age of 61.73 years in Iran, which also agrees with our findings. The present study revealed a male:female ratio of approximately 3:1 (75.8% vs. 24%). Similarly, Teimori et al. [3] reported a male:female ratio of 2.9:1, while Rozman and Montserrat [21] also reported that patients with CLL were more likely to be male.
In the present study, patients with del 17p presented at an advanced clinical stage (i.e., Binet stage C), whereas Döhner et al. [13] reported that most of their patients presented at Binet stage B. However, Döhner et al. [13] reported higher LDH levels among patients with del 17p, which agrees with our findings of elevated LDH levels in this group of patients. We failed to detect a significant association between del 17p and CD38 positivity (P=0.9), which agrees with the previously reported findings of Lin et al. [22]. These findings confirm that del 17p is an independent prognostic marker for CLL.
Our findings generally agree with the results of other local studies, although differences can be observed when our results are compared to those from other global regions. These differences may be attributable to geographical and/or ethnic differences in disease biology and genetic factors. For example, during a 24-month follow-up period, 70% of our patients required treatment, while Delgado et al. [20] reported that 52% of their patients required treatment. Furthermore, our median time from diagnosis to starting treatment was 11 months (range, 0–24 mo), while Strati et al. [12] have reported an interval of 15 months. These differences may be related to Pakistan being a developing country, which may limit patients' access to tertiary medical facilities and lead to their disease being diagnosed at a relatively advanced stage. In the present study, the overall response rate was 64.3% (N=9), although 5 patients (35.7%) were refractory to primary therapy, and the 2-year EFS rate was 11% with a median interval of 9 months (range, 1–24 mo). In contrast, Strati et al. [12] reported a median EFS interval of 14 months. In our study, prolonged EFS was associated with the absence of B symptoms, Binet stage A–B, del 17p clone size of <25%, and B2 microglobulin of <4 mg/L, but not with age, CD38, or ZAP70. Strati et al. [12] have reported that prolonged EFS was associated with an age of <65 years and a del 17p clone size of >50%, but not with clinical stage, CD38, or ZAP70. In our study, the 2-year OS rate was 65%, and poor OS was associated with B symptoms (P=0.04) but not with age, gender, Binet stage, del 17p clone size, CD38, or ZAP70. Delgado et al. [20] have reported that poor OS was associated with Binet stage C, a B2 microglobulin concentration of >2× the upper normal limit, and a higher percentage of cells with del 17p. Nevertheless, further multicenter studies with larger samples of patients are needed to better estimate the prevalence of this deletion and the related outcomes.
In conclusion, the frequency of del 17p was relatively high among patients with CLL who visited our center in Pakistan. However, not all of these patients required treatment. The overall response rate was 64% for conventional first-line therapy, although this was associated with a short EFS interval (median, 9 mo). Nevertheless, the introduction of promising novel drugs may help improve the response rate among this high-risk subgroup and subsequently prolong their EFS and OS intervals.
 XML Download
XML Download