

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Inborn errors of bile acid synthesis (IEBAS) are categorized as genetic metabolic disorders that usually manifest as prolonged jaundice in the neonatal period. They are caused by enzyme defects involving the pathways for bile acid synthesis from cholesterol in the liver.1 With at least 17 enzymes involved, 9 types of IEBAS have been identified to date.23 As their clinical manifestations may range from liver failure in early infancy to cirrhosis or progressive neuropathy in adolescence or adulthood, early recognition and diagnosis is crucial to prevent fatal outcomes.12
Oxysterol 7α-hydroxylase deficiency, one of the IEBAS, is considered to have the least prevalence among the known IEBAS.4 Since only 4 cases have been reported to date, clinical manifestations or prognoses have not been clearly established.
We describe the first Korean case of IEBAS in an infant showing rapid progression of liver failure, who completely recovered after undergoing liver transplantation (LT).
Go to : 
CASE DESCRIPTION
A 3-month-old boy was referred to our tertiary center for evaluation of progressive cholestatic hepatitis in December 2015. He was born at 39 weeks of gestation by vaginal delivery, weighing 3.4 kg and without perinatal problems, at a local hospital. His jaundice was noticed about 1 week after birth. During the follow-up of his prolonged jaundice at a nearby hospital, he underwent ultrasonography of the abdomen, scintigraphy, and liver biopsy at about 1 month of age, which excluded biliary atresia.
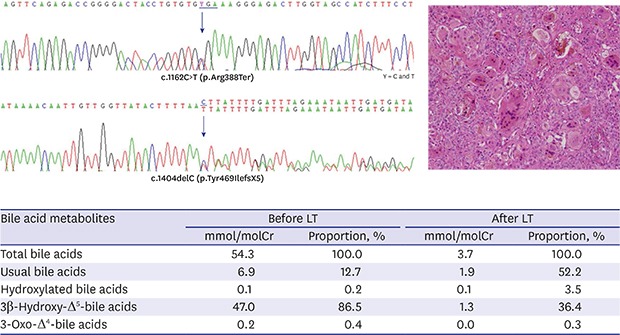
On admission, he was active and had normal growth with a weight of 7.3 kg (85–90th percentile) and height of 62.3 cm (50–75th percentile). Physical examination showed yellowish skin color, icteric sclera, and hepatosplenomegaly. His development was appropriate for his age, with head control. His blood tests showed the following: white blood cell count, 10,200/mm3; hemoglobin level, 10.0 g/dL; and platelet count, 180,000/mm3. Liver function test results were as follows: aspartate aminotransferase/alanine aminotransferase levels, 1,599/301 IU/L; γ-glutamyltransferase (GGT) level, 22 IU/L; alkaline phosphatase level, 1,423 IU/L; total bilirubin/direct bilirubin levels, 9.3/7.0 mg/dL; prothrombin time, 2.74 INR; activated partial thromboplastin time, 74.3 seconds; protein/albumin levels, 5.0/3.1 g/dL; and cholesterol level, 54 mg/dL. The serum bile acid level was increased to 131.4 µmol/L. The serum 25-OH vitamin D3 level was 6.7 ng/mL. Renal function and electrolyte levels were normal. Titers for toxoplasmosis, herpes, rubella, and cytomegalovirus were negative. Liver biopsy revealed giant cell change of the hepatocytes and moderate portal inflammation with periportal fibrosis (Fig. 1A). Immunohistochemical staining showed that the expression of bile salt export protein (BSEP) was focally decreased but not completely absent (Fig. 1B). Additionally, CD10 expression was not observed.
 | Fig. 1Histologic findings of the liver biopsy. (A) Giant cell transformation of hepatocytes is observed (arrow). There is moderate portal inflammation and periportal fibrosis with extramedullary hematopoiesis (hematoxylin and eosin stain, × 200). (B) Immunohistochemical assay shows that the expression of bile salt export protein is focally decreased but not completely absent.
|
Under the suspicion of IEBAS, a urine sample was collected and dropped onto a filter paper, making dried urine spots, which was sent to a Japanese laboratory center. Gas chromatography/mass spectrometry analysis was performed for evaluation of bile acid metabolites, as described previously.5 It revealed profuse urinary excretion of unusual bile acids, 3β-monohydroxy-Δ5-bile acid derivatives, which account for more than 86.5% of total bile acids apart from ursodeoxycholic acid (UDCA) (Table 1). We confirmed the diagnosis of oxysterol 7α-hydroxylase deficiency.
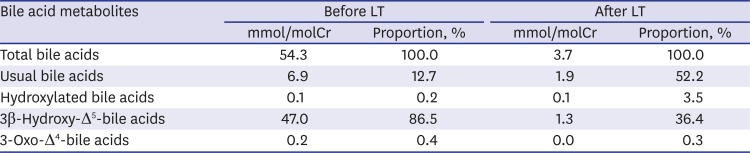
Table 1
Results of the analysis of bile acids in dried urine spots on filter paper by GC/MS before and after LT
![]()
Genetic analysis by direct sequencing of 6 exon-coding lesions in the CYP7B1 gene identified a compound heterozygote mutation. One was a nonsense mutation, p.Arg388Ter, in exon 5, and the other was a frameshift mutation, p.Tyr469IlefsX5, in exon 6 (Fig. 2).
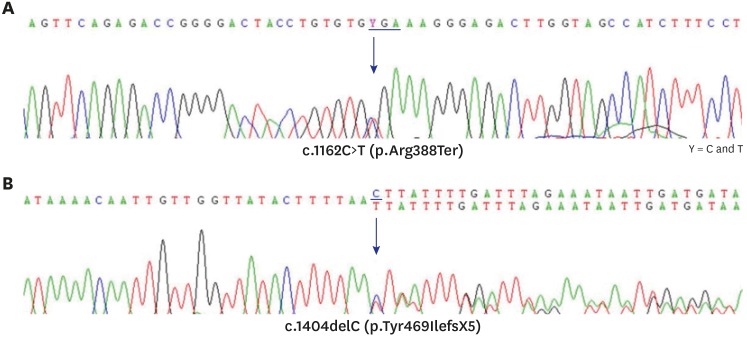 | Fig. 2Result of Sanger sequencing for exons 5 and 6 of the CPY7B1 gene. (A) There is a substitution of C to T at nucleotide 1,162 in exon 5 that changes arginine to a stop codon at amino acid position 388. (B) A deletion of C at nucleotide 1,404 in exon 6 makes a frame shifting change inducing tyrosine as the first changed amino acid at codon 469 and ending in the new reading frame in a stop at position 5.
|
Although the patient received standard supportive care for cholestatic diseases with fat-soluble vitamins and UDCA (15 mg/kg/day), his liver function deteriorated. As oral primary bile acids were unavailable in Korea, additional medication was not administered even after diagnosis. Eventually, he received living donor transplantation from his mother because of the rapid progression of liver failure.
Follow-up urinary bile acid analysis showed markedly decreased unusual bile acid metabolites after LT (Table 1). His liver function has been maintained within the normal range. He had normal growth with a weight of 16.1 kg (90th percentile) and showed normal developmental milestones until the current follow-up at 33 months of age.
Go to :
DISCUSSION
We describe the first case of oxysterol 7α-hydroxylase deficiency in Korea, diagnosed by mass spectrometric analysis of urinary bile acids, an unavailable technique in Korea and confirmed by CYP7B1 mutation. Our patient was the youngest reported patient to undergo successful living donor liver transplantation (LDLT) for oxysterol 7α-hydroxylase deficiency to date.
The synthesis of primary bile acids, cholic acid (CA) and chenodeoxycholic acid (CDCA) occurs in the liver via 2 main pathways: the neutral (classic) pathway and the acidic (alternative) pathway.134 Oxysterol 7α-hydroxylase is involved in the conversion of 3β-hydroxy-5-cholestenoic acid to 3β,7α-dihydroxy-5-cholestenoic acid in an essential step of the acidic pathway that is particularly relevant in the first year of life. In oxysterol 7α-hydroxylase deficiency, atypical sterol intermediates such as 3β-hydroxy-5-cholenoic acid are more likely to be produced especially during infancy, which have cholestatic and hepatotoxic properties.2 This may explain the clinical features of early liver failure in oxysterol 7α-hydroxylase deficiency. In the brain, an extrahepatic tissue, oxysterol 7α-hydroxylase is involved in cholesterol degradation and neurosteroid metabolism as a modification of dehydroepiandrosterone, where the loss of functions may cause neurologic manifestations.26
Oxysterol 7α-hydroxylase deficiency is an autosomal recessive disorder caused by mutations in the CYP7B1 gene containing 6 exons on chromosome 8q21.3.7 Recently, mutations in the CYP7B1 gene have been shown in patients with hereditary spastic paraplegia type 5 (HSP5), which manifests with lower-extremity weakness and spasticity, posterior column sensory impairment, and bladder dysfunction with an onset age of 1 to 40 years.26 The different manifestations of the diseases caused by mutations in one gene, CYP7B1, have explained the differences in the CYP7B1 substrates accumulated, i.e., different substrates build up in different presentations.8 Among the 17 identified CYP7B1 mutations associated with HSP5, some loci were found in subjects with oxysterol 7α-hydroxylase deficiency; one of them was also found in our case.8 It is necessary to monitor the development of neurologic symptoms in patients with oxysterol 7α-hydroxylase deficiency although abnormal findings do not appear on presentation.
IEBAS are usually suspected when patients present with cholestasis and a normal serum GGT level, normal or low serum levels of total bile acid, or rapid progression to end-stage liver disease even in early infancy.1 We first suspected our patient to have progressive familial intrahepatic cholestasis (PFIC) type 1 or type 2, which is biochemically characterized with cholestasis with normal serum GGT and high serum bile acid concentration in infancy. However, diagnosis of PFIC types 1 or 2 could be excluded with immunohistochemical staining of liver biopsy when BSEP expression was noted with deficiency of canalicular ectoenzyme CD10 expression. High level of serum total bile acid in our patient was considered to be associated with UDCA administered prior to referral. To reduce the possibility of misinterpretation, it is suggested that specimens be collected 5 to 7 days after UDCA discontinuation for the diagnosis of IEBAS.
The recommended screening method for IEBAS is mass spectrometry, which can detect excessive amounts of atypical bile acids in urine or serum as a consequence of enzyme deficiency.12 When we suspected our patient of having a type of IEBAS after excluding other etiologies for his early liver failure, there was no available laboratory in Korea to perform the analytical technique. Therefore, we sent a dry urine sample to a center in Japan, where the second and third reported cases of oxysterol 7α-hydroxylase deficiency were diagnosed.910 According to a recent survey in 21 European countries, physicians reported that the main obstacles to diagnosing the IEBAS were the lack of available specialized laboratories, followed by the lack of or limited experience with the diseases.11 In fact, only 10 centers in 9 European countries have expert laboratories that perform mass spectrometry for diagnosis.
In most other IEBAS, primary bile acid replacement has been considered responsible, and currently, oral CA is suggested as an effective therapy.12 Since the first case of oxysterol 7α-hydroxylase deficiency was described in 1998, 3 more cases have been reported to date.9101314 Based on this limited experience, oxysterol 7α-hydroxylase deficiency has shown relatively poor prognosis compared with other IEBAS. It showed worsening with UDCA and no improvement with oral CA, requiring LT in 3 cases, while one patient was successfully treated with oral CDCA without transplantation in a recent study.14 We reviewed 4 reported cases and have summarized the clinical findings in Table 2.
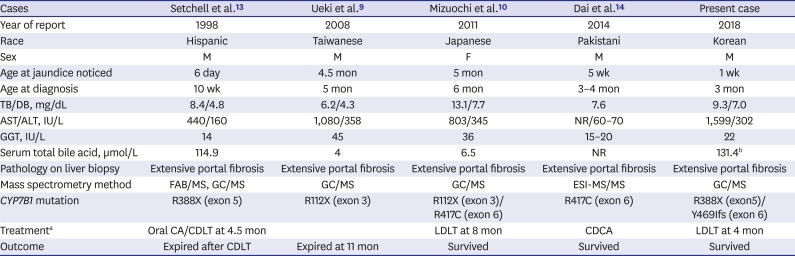
Table 2
Clinical characteristics of the reported cases with oxysterol 7α-hydroxylase deficiency
| Cases | Setchell et al.13 | Ueki et al.9 | Mizuochi et al.10 | Dai et al.14 | Present case |
|---|---|---|---|---|---|
| Year of report | 1998 | 2008 | 2011 | 2014 | 2018 |
| Race | Hispanic | Taiwanese | Japanese | Pakistani | Korean |
| Sex | M | M | F | M | M |
| Age at jaundice noticed | 6 day | 4.5 mon | 5 mon | 5 wk | 1 wk |
| Age at diagnosis | 10 wk | 5 mon | 6 mon | 3–4 mon | 3 mon |
| TB/DB, mg/dL | 8.4/4.8 | 6.2/4.3 | 13.1/7.7 | 7.6 | 9.3/7.0 |
| AST/ALT, IU/L | 440/160 | 1,080/358 | 803/345 | NR/60–70 | 1,599/302 |
| GGT, IU/L | 14 | 45 | 36 | 15–20 | 22 |
| Serum total bile acid, µmol/L | 114.9 | 4 | 6.5 | NR | 131.4b |
| Pathology on liver biopsy | Extensive portal fibrosis | Extensive portal fibrosis | Extensive portal fibrosis | Extensive portal fibrosis | Extensive portal fibrosis |
| Mass spectrometry method | FAB/MS, GC/MS | GC/MS | GC/MS | ESI-MS/MS | GC/MS |
| CYP7B1 mutation | R388X (exon 5) | R112X (exon 3) | R112X (exon 3)/R417C (exon 6) | R417C (exon 6) | R388X (exon5)/Y469Ifs (exon 6) |
| Treatmenta | Oral CA/CDLT at 4.5 mon | LDLT at 8 mon | CDCA | LDLT at 4 mon | |
| Outcome | Expired after CDLT | Expired at 11 mon | Survived | Survived | Survived |
M = male, F = female, TB = total bilirubin, DB = direct bilirubin, AST = aspartate aminotransferase, ALT = alanine aminotransferase, NR = not reported, GGT = γ-glutamyltransferase, FAB/MS = fast atom bombardment ionization/mass spectrometry, GC/MS = gas chromatography/mass spectrometry, ESI-MS/MS = electrospray ionization tandem mass spectrometry, CDCA = chenodeoxycholic acid, CA = cholic acid, CDLT = cadaveric donor liver transplantation, LDLT = living donor liver transplantation, UDCA = ursodeoxycholic acid.
aTreatment except for usual supportive care with fat-soluble vitamins or UDCA; bThe level was checked during administration of UDCA (in text).
![]()
In metabolic liver disease, LT is an established therapeutic option. In transplantation, haplo-matched donors as first degree-related donors are considered to be potentially heterozygous carriers, possibly causing recurrence of the original disease. However, there have been reports that haplo-matched donor grafts have been safely used in this and other inherited metabolic liver diseases without negative impact on recipients.1516 In our case with autosomal recessive trait, a haplo-matched donor graft from the mother was successfully transplanted, and urinary bile acid analysis after transplantation showed no recurrence of the original disease. This was also confirmed in a report from Japan.10
We suggest that IEBAS should be differentiated in neonates with prolonged jaundice especially when progressive liver failure is observed and the etiology is unclear. For early diagnosis, urine bile acid analysis should be performed foremost in patients with persistent cholestasis and a normal serum GGT level regardless of the bile acid concentration. In cases of oxysterol 7α-hydroxylase deficiency, LT may be inevitable to prevent mortality due to the rapid progression of liver failure.
Go to :
 XML Download
XML Download