

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Bone is a frequent site for primary tumor cells to spread and more than 600,000 people every year suffer from bone metastasis (BM) in the US.[1] Breast, prostate and lung cancers commonly lead to BM and more than 70% of patients with breast cancer (BC) and BM have a high rate of morbidity and mortality.[234] Patients with BM may suffer from a variety of skeletal-related complications (SRCs) such as bone pain, pathologic fractures, spinal cord compression, and difficulty to walk.[5] Additionally, once a patient with BM experiences a first SRC, the likelihood of experiencing a second SRC is greatly increased.[5] Studies regarding normal physiology of bone metabolism, interactions between bone and advanced cancer, and genetics related with BM have been performed in the laboratory.[678] In addition, surgeries, radiation therapy and chemical therapy using target agents are performed in the clinical field.[910] However, despite the multidisciplinary approach, a large proportion of BM remains incurable and treatment strategies for BM usually have a palliative effect.[6] The decrease in effectiveness is due to fact that the pathophysiology of BM from BC is a multistep and complex process that consists of escapes of tumor cell from primary site, a crosstalk between disseminated BC cells and bone-derived molecules, leading to bone and reconstitution of secondary tumors at the bone.[1112] Although novel therapies which target the pathways involved in BM are emerging, research which focuses on identifying key upstream regulators related with the molecular signaling pathway of BM remains clinically relevant to the prevention of BM.[11] Using statistical machine learning methods can help us find key upstream regulators from a causal network model inferred from clinical, genomic and environmental data related with BM. In turn, this can improve clinically preventive practices and reduce the adverse effect related with target therapy of BM.
Application of machine learning methods have been widely used to get the statistical relationship and causal networks from large and complicated health data.[1314] As one of machine learning methods, causal Bayesian networks (CBN) have been used to learn causal network inferred from known collected genomic data.[1516] It has been shown that the gene expression patterns could be influenced by multistep complex processes from primary site, through dissemination, into metastasis.[717] Therefore, the relationships of 1 or 2 genes or molecules cannot reflect how combination of many genes' expressions changes according to surrounding environment. We need to seek for the relationships from interactions from network of genes and further search for plausible causal interactions. Although studies have presented the gene changes according to BM of BC (BMBC), none of the studies developed research to build an integrated causal network consisting of gene signatures associated to BMBC.[17] The CBN analysis of dataset of the expressed genes in BMBC can provide insight into the causal relationship and upstream regulators governing BMBC. There are 3 specific niches in homing of tumor cells into bone: the endosteal niche, the haematopoietic stem cell niche and the vascular niche.[18] The endosteal niche related with osteoblast has a key role in the homing and adhesion of circulating tumor cells into bone and the clinically relevant and important network related with BM seems to be in the endosteal niche.[1819] Therefore, we conducted a CBN analysis of microarray data from the Gene Expression Omnibus (GEO) to get a causal gene expression network consisting of genes related with BMBC and osteoblast for identifying the upstream regulators of BMBC.
METHODS
After collecting gene expression data from GEO following inclusion criteria, we cleaned, normalized, and discretized the gene expression levels. Then, we selected candidate genes from the list of genes that were common to the studies involving BMBC and osteoblast. In the next step, we learned CBN structure by combining the gene expression data with published literature and knowledge. We further learned CBN parameters (conditional probabilities) and evaluated the final model to see how well it fits the data (maximum likelihood and conditional independencies), compared it with current knowledge (published literature articles) and how well it predicts future instances (receiver operating characteristic [ROC] curve and relative risk [RR]) (Fig. 1).
We further describe each step that we summarized above in more detail:
1. Collecting data from GEO
We have retrieved microarray datasets from the GEO database of the National Center for Biotechnology Information (NCBI) of National Institute of Health (https://www.ncbi.nlm.nih.gov/geo/, GEO, NCBI accessed in November 2017).[20] We have used the following inclusion criteria: (1) datasets with the GEO series (GSE) of BMBC, BC without metastasis and osteoblast; (2) studies should be measuring gene expression of human (homo sapiens); (3) studies tissue extracted from metastatic bone and the cancerous region of breast and experimental human cell lines or normal bone tissue for osteoblast; and (4) BC without metastasis was diagnosed as breast ductal carcinoma without metastasis when the tissues were extracted by biopsy or surgery. As a result, there were seven studies that were identified for this study (ten GSEs and 48 GEO sample) (Table 1).
2. Data mining and selection of candidate genes
In this section, we describe how we prepared the datasets that were collected from GEO and in the next section we describe how we learn causal network of genes commonly expressed in BMBC and osteoblasts using CBN.
1) Data cleaning, averaging, normalization and discretization (CANDi)
Because gene symbols in the GSEs (set of samples) are written by gene Identifier (ID) not gene name, we changed gene IDs in GSE into corresponding gene symbol in matched GPL (technology platform of microarray analysis used in study) (Cleaning). Then, if single gene had several raw expression values of gene, we transformed the raw values of each gene into a mean value of the raw values (Averaging). For meaningful comparison of gene expression level, the relative data from individual studies should be transformed into normalized data. We normalized the averaging data into z-scores and performed the normalization task on study-by-study basis (Normalization).[21] Bayesian Network Inference with Java Objects (Banjo) is one of computational modeling tools based on data-driven method and Banjo utilize Bayesian network frameworks to result in directed inference network.[222] Because Banjo implement Baysian Dirichlet equivalence scoring metric of data consisting of discrete variables, we transformed the normalized data to discrete values (Discretization). For each gene, all expressions z score values (z) were discretized as less than −1 (z<−1), between −1 and 1 (−1≤z≤1), and over 1 (z>1) were discretized as low, no change, and high, respectively. We sequentially performed the same CANDi process per each study.
2) Selection of candidate genes
We selected common genes there were all presented in GSE of osteoblast, normal bone tissue and BMBC. Among the common genes, if the genes simultaneously expressed in BC without metastasis such as genes expressed in BC in-situ, we excluded the genes in the study. As a result, ten GSEs (5 for osteoblast and 5 for BMBC) from seven studies and 1,218 genes were selected (Table 1). After collection of all data in ten GSEs, we prepared a dataset (denote as D°) that consists of variables that represent expression levels (low, no change, and high) of 1,218 genes and a variable called group that represents whether a subject has osteoblast or BM. In the final dataset D°, there were 48 subjects (13 GSMs with osteoblast and 35 GSMs with BMv) (Table 1).
3. Learning CBN structure
The structure of CBN is directed acyclic graph (DAG) consisting of nodes and intervening arrows.[13] The arrows in DAG are interpreted in terms of probabilistic conditional independence between nodes.[23] First degree Markov Blanket (MB) of a variable X in CBN (denote as MB [X]) is defined as set of variables that is the direct causes (parents) of X and direct effects (children) of X and direct causes (parents) of direct effects (children) of X (of course, X itself is excluded from MB [X]). Second degree MB of X, third degree MB of X, etc., can be defined as MB (MB [X]), MB (MB [MB (X)], etc., respectively.
An example CBN structure is shown in Figure 2. In that structure, for example, the expression of NFKB2 influences the likelihood of the presence of BM which influences on expressions of MAP2K2 and TSO. In parallel, conditional independence between nodes shows as the probability about expression of MAP2K2 or TSO is not influenced by NFKB2 given information of BM. Also in Figure 2, first degree MB of MAP2K2, MB (MAP2K2), is {BM, TSO} and second degree MB of MAP2K2, MB (MB [MAP2K2]), is {NFKB2, BM, TSO}.
We used Banjo,[24] a Bayesian network learning tool, to learn 2 CBNs, i.e., a CBN with all 1,218 candidate genes from all the studies and a CBN with 70 BC-Relevant Genes (BCRGs).
1) CBN with candidate genes
To learn the CBN with all 1,218 candidate genes from all the studies, we ran 18 independent runs of Banjo (3 independent 1 hr, 3 hr, 6 hr, 12 hr, 24 hr, and 36 hr runs; total of 246 hr runs) with the dataset D° and by limiting maximum parents to be 5. Among 18 best log likelihood structures that were reported by each independent Banjo run, we chose the network with the highest log likelihood (denoted as S°; note that S° includes 1,219–1,218 variables, each representing a gene expression and a variable named group, representing whether a subject has osteoblast or BM).
2) CBN with BCRGs
CluePedia, one of Cytoscape plugins, have been known as a provider for pathways, processes or disease related with gene, miRNA and protein in conjunction with ClueGo.[25] Using ClueGo and CluePedia Cytoscape plugin, we found 13 BCRGs (that represents genes having more close association to disease and acceptable prior knowledge) registered in kyoto encyclopedia of genes and genomes (KEGG).[26] To further analyze possible interactions of 13 BCRGs and 57 genes from second degree MB of group node in S°, we again ran Banjo with a new dataset (denote as D◆) that combines the expressions of these 70 genes (13 BCRGs and 57 genes from second degree MB). The condition of setting files for the second Banjo analysis was same as previous condition except that we used D◆ with data of 71 variables extracted from data of 1,219 variables in D° (48 subjects). Among 18 best log likelihood structures that were reported by each independent Banjo run, we chose the network with the highest log likelihood (denoted as S◆; note that S◆ includes 71–70 variables, each representing a gene expression and a variable named group, representing whether a subject has osteoblast or BM).
4. Learning causal Bayesian parameters
In the network with the highest log likelihood in the second Banjo analysis (S◆), we represent first degree MB of the group variable using Bayesian network graphical network interface (GeNIe; version 2.2.1; BayesFusion, Pittsburgh, PA, USA) to learn parameters (probabilities). Learning the parameters of the Bayesian structure was implemented using the data set with the dataset that was extracted from the original dataset (48 observations). In addition, we investigated the relationships among nodes and the influence of status of BMBC on expressions of other genes in the Bayesian structure which comprised of 34 nodes within MB (group) of S◆ (denoted as S*; note that S* includes 34–33 variables, each representing a gene expression and a variable named group, representing whether a subject has osteoblast or BM).
5. Validations
We further validated CBN structure S* by how well S* fits the data (maximum likelihood and conditional independencies), compared S* with current knowledge (published literature articles) and how well S* predicts future instances (ROC and RR). We searched for orders of variables with best probabilistic score using Markov chain over Monte Carlo (MCMC) simulation and compared the result with the order of nodes in Bayesian structure (call this order analysis).[27] The variables we used were direct cause (parent) and direct effect (children) genes and disease node that showed the strongest influence through conditional independency test (we refer these variables as Validation Variables and denote as V*).[28] We performed order analysis with 3 independent 1 hour runs and we compare the best order with S* and current knowledge. Also we investigated degree of conditional independencies among variables in CBN structure S* and check how well conditional independency relationships (d-separation and d-connectivity [29]) among variables. We also evaluated CBN structure S* using leave one out cross test and the area under ROC (AUROC) using GeNIe. We obtained the prediction rate of BMBC of CBN structure S* with the final dataset that includes the same number of variables in S*,i.e., 34–33 variables, each representing a gene expression and a variable named group, representing whether a subject has osteoblast or BM, and 48 subjects. We calculate how much we can expect BM with the final dataset of BM if we have the information in Validation Variables V*. We investigated the information of genes within in CBN structure S* whether the structure reflected the current knowledge and previous studies using Cytoscape 3.6 [26] and searching articles in PubMed in NCBI (www.ncbi.nlm.nih.gov/pubmed).
In final process of validation, we investigated all joint distributions of genes in Validation Variables V* given BMBC and also how all possible combination of gene expression patterns predict BMBC and evaluated the minimum combination with maximum RR of combined gene expression of the genes in the final model and significant genes. We have used 12 genes in Validation Variables V* and used CBN S* and calculated all 16,777,215 (=∑12i=1(12i)3i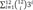 =(3+1)12−1=412−1) different gene configurations g with the collected dataset and using SMILE library (https://www.bayesfusion.com/smile-engine) and C++ program and calculate the following:
=(3+1)12−1=412−1) different gene configurations g with the collected dataset and using SMILE library (https://www.bayesfusion.com/smile-engine) and C++ program and calculate the following:
=(3+1)12−1=412−1) different gene configurations g with the collected dataset and using SMILE library (https://www.bayesfusion.com/smile-engine) and C++ program and calculate the following:where D represents a subject has BMBC and V is any subset of V*. Among the gene configurations g that predicts BMBC with high or low probability (i.e., P(D|V=g)>0.99999 or P(D|V=g)<1.0×10−6).
To find the minimum set of combination of gene expression patterns that give us a maximum RR, we have calculated the following:
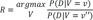
where, V represents any subset of V*, R represents a set of the minimum number of genes that maximizes the RR term, v=argmaxgP(D|V=g)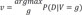 and v′=argmingP(D|V=g)
and v′=argmingP(D|V=g)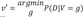 . Note that v and v′ represents 2 different gene expression patterns among the genes in R that maximizes and minimizes P(D|V) respectively. We report the top 4 RR that we calculated from the dataset.
. Note that v and v′ represents 2 different gene expression patterns among the genes in R that maximizes and minimizes P(D|V) respectively. We report the top 4 RR that we calculated from the dataset.
and v′=argmingP(D|V=g). Note that v and v′ represents 2 different gene expression patterns among the genes in R that maximizes and minimizes P(D|V) respectively. We report the top 4 RR that we calculated from the dataset.RESULTS
We report the 3 CBN structures, i.e., a CBN with candidate genes (S°), a CBN with BCRGs (S◆), and a CBN as the first degree MB of group variable in S◆(S*) described in the method section. We also report results of their validations.
1. CBN with common genes of all studies
Among the 18 CBNs, the CBN with the best log likelihood score reported by Banjo (i.e., log P(D° | S°)=−49,091.645 where S° a CBN with 1,219 variables and D° is the dataset with the same number of variables with 48 subjects) was significantly better fitting the data than the second best CBN (i.e., P(D°|S°)P(D°|S°)+P(D°|S′)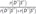 )>99.999% where S′ is the second best CBN). S° includes 1,218 gene expression and a variable called group (representing whether a subject has osteoblast or BM) (Fig. 3). Among 1,219 variables, we selected 59 genes within second degree MB of group (Fig. 4). In addition, CluePedia, one of Cytoscape plugins, identified 13 BCRGs that had common pathways - mammalian target of rapamycin signaling pathway, ErbB signaling pathway, signaling pathways regulating pluripotency of stem cell - in prostate cancers, thyroid cancer and non-small cell lung cancer (Fig. 5).
)>99.999% where S′ is the second best CBN). S° includes 1,218 gene expression and a variable called group (representing whether a subject has osteoblast or BM) (Fig. 3). Among 1,219 variables, we selected 59 genes within second degree MB of group (Fig. 4). In addition, CluePedia, one of Cytoscape plugins, identified 13 BCRGs that had common pathways - mammalian target of rapamycin signaling pathway, ErbB signaling pathway, signaling pathways regulating pluripotency of stem cell - in prostate cancers, thyroid cancer and non-small cell lung cancer (Fig. 5).
)>99.999% where S′ is the second best CBN). S° includes 1,218 gene expression and a variable called group (representing whether a subject has osteoblast or BM) (Fig. 3). Among 1,219 variables, we selected 59 genes within second degree MB of group (Fig. 4). In addition, CluePedia, one of Cytoscape plugins, identified 13 BCRGs that had common pathways - mammalian target of rapamycin signaling pathway, ErbB signaling pathway, signaling pathways regulating pluripotency of stem cell - in prostate cancers, thyroid cancer and non-small cell lung cancer (Fig. 5).2. CBN with BCRGs
Among the 18 CBNs, the CBN with the best log likelihood score reported by Banjo (i.e., log P(D◆|S◆)=−2,613.829 where S◆ a CBN with 71 variables and D◆ is the dataset with the same number of variables with 48 subjects) was significantly better fitting the data than the second best CBN (i.e., P(D◆|S◆)P(D◆|S◆)+P(D◆|S′) >99.999% where S′ is the second best CBN) (Fig. 6). The first degree MB of group variable was selected from S◆ (we denoted this CBN with 34 variables as S*): 3 genes that were direct causes (parents) of group (NFKB2, UBIAD1, and HEBP1), 12 genes that were direct effects (children) of group (FZD1, MAP2K2, BTNL8, TSPO, HOXB2, FOLH1, KIF11, SLC10A3, PSAT1, CACYBP, S100PBP, and ZFP36L2) and 20 genes that were direct causes (parents) of direct effects (children) of group (BTNL8, TEP1, L1TD1, TSPO, PSMB8, RPS6KB2, DLL3, TNFRSF11B, MAPK3, FASTKD3, ZNF273, PPP2CA, AGA, IFI30, ZDHHC6, INPP1, YWHAB, EPS8, RANBP6, and TATDN2).
>99.999% where S′ is the second best CBN) (Fig. 6). The first degree MB of group variable was selected from S◆ (we denoted this CBN with 34 variables as S*): 3 genes that were direct causes (parents) of group (NFKB2, UBIAD1, and HEBP1), 12 genes that were direct effects (children) of group (FZD1, MAP2K2, BTNL8, TSPO, HOXB2, FOLH1, KIF11, SLC10A3, PSAT1, CACYBP, S100PBP, and ZFP36L2) and 20 genes that were direct causes (parents) of direct effects (children) of group (BTNL8, TEP1, L1TD1, TSPO, PSMB8, RPS6KB2, DLL3, TNFRSF11B, MAPK3, FASTKD3, ZNF273, PPP2CA, AGA, IFI30, ZDHHC6, INPP1, YWHAB, EPS8, RANBP6, and TATDN2).
>99.999% where S′ is the second best CBN) (Fig. 6). The first degree MB of group variable was selected from S◆ (we denoted this CBN with 34 variables as S*): 3 genes that were direct causes (parents) of group (NFKB2, UBIAD1, and HEBP1), 12 genes that were direct effects (children) of group (FZD1, MAP2K2, BTNL8, TSPO, HOXB2, FOLH1, KIF11, SLC10A3, PSAT1, CACYBP, S100PBP, and ZFP36L2) and 20 genes that were direct causes (parents) of direct effects (children) of group (BTNL8, TEP1, L1TD1, TSPO, PSMB8, RPS6KB2, DLL3, TNFRSF11B, MAPK3, FASTKD3, ZNF273, PPP2CA, AGA, IFI30, ZDHHC6, INPP1, YWHAB, EPS8, RANBP6, and TATDN2).3. Learning causal Bayesian parameters
Probabilities (parameters) of the CBN with 34 variables that represents the first degree MB of group variable (S*) of CBN with BCRGs (S◆) was learned from a new dataset (denoted as D*) with data of 34 variables extracted from data of 71 variables in D◆ (48 subjects) (Fig. 7). The NFKB2 gene in S* (direct cause [parent] of the group variable) was registered as BCRGs in the KEGG and NF-κB protein encoded by the NFKB2 gene is known as transcription factor promoting BC stem cell.[30] Among 12 direct effects (children) genes of BMBC (group variable) in S*, 5 genes (SLC10A3, FZD1, BTNL8, KIF11, and CACYBP) were reported as the genes having close association with chemotherapy resistance.[3132333435] In addition, KIF11, MAP2K2, PSAT1, TSPO, and ZFP36L2 genes within direct effects (children) of BMBC (group variable) were known as having relationship with cancer progression and metastasis.[36373839]
According to parameters learned in S*, comparing a subject that has BMBC (group variable is in state 1, i.e., BM) with a subject that has osteoblast, among 15 genes that showed expression pattern change, FOLH1 and ZFP36L2 were the most significantly changed genes (FOLH1 expression changed from no change [state 1] to low [state 0] and ZFP36L2 expression changed from high [state 2] to no change [state 1]) (Fig. 8).
4. Assessment and validations
We further evaluated prediction performance of the CBN S* parameterized by D* with leave one out cross validation test achieved 68.38% (1,116 correct predictions out of 34×48 cases). When we used only direct causes (parents) and effects (children) of BMBC (group variable) in S* (i.e., 16 variables out of 34 variables) leave one out cross validation test retried 82.16% (631 correct predictions out of 16×48 cases). AUROC predicting BMBC and osteoblast were 1 in S* with the 16 variables (direct causes and effects of BMBC). Also in the CBN S* parameterized by D*, just using the 3 direct causes of BMBC (group variable) the probability of a subject having BM were over 90% (Table 2).
We have also calculated how likely any 2 variables are conditional independent (note that we are calculating the probabilities of conditional independencies, so the lower the probability is the higher the chances the variables are not independent) among the 16 variables (direct causes and effects of BMBC [group variable]) and group variable.[28] Calculated probabilities of conditional independence showed that conditional independency relationships (d-separation and d-connectivity [29]) among 16 variables and group variable in the S* were agreeing with the structure of S* (Fig. 9). These probabilities of conditional independence allowed us to further select 12 genes (Table 3) that showed higher relationships with BMBC (group variable). Figure 6 shows that except 1 gene, all 13 BCRGs are connected to BMBC (group variable) in CBN with BCRGs. Among the 12 genes, 6 genes (MAPK2, MAPK3, NFKB2, FZD1, DLL3, and RPS6KB2) were variables in S*. We report 4 maximum RRs of combined gene expression patterns (Table 3). Table 3 shows UBIAD1, HEBP1, BTNL8, TSPO, PSAT1 and ZFP36L2 genes consistently appears in the highest RRs combined gene expression patterns. The difference of expression levels of ZFP36L2 was especially significant because its expression differed from low (represented as 0) to high (represented as 2) in all combination patterns whereas most of the other genes differed to only no change (represented as 1).
Searching through variable orders in the CBN (if variable A is (indirect) cause of variable B, then A is said to be in the higher order of B) has been suggested to be a different way of searching for best fitting CBN.[27] Using the 16 variables (direct causes and effects of BMBC [group variable]) the order with the best score was in the following order (higher to lower): NFKB2, HEBP1, group, FOLH1, ZFP36L2, HOXB2, PSAT1, TSPO, CACYBP, MAP2K2, UBIAD1, BTNL8, KIF11, S100PBP, and FZD1. Two direct cause (parent) genes (NFKB2 and HEBP1) of group variable in S◆ (Fig. 6) consistently appeared in the higher order than group variable. However, UBIAD1 gene that was the direct cause (parent) of group variable in S◆, appeared in the lower order than group variable. The genes with the highest change in probability to be expressed high (State 2) given the fact that a subject has BMBC (FOLH1 and ZFP36L2), appeared right next to group variable in the order. The best CBN structure that was identified by the order analysis was significantly better in terms of log likelihood, however, the conditional independencies of the 16 variables (direct causes and effects of BMBC [group variable]) were consistent with S*.
DISCUSSION
We have built a causal inference statistical model using a machine learning tool, i.e., CBN, from GEO gene expression data of osteoblast and BMBC. While building the causal inference statistical model, we have also incorporated medical and biological prior knowledge such as clinical medical practice and information of BCRGs registered in KEGG. We also validated the causal inference statistical model using data, statistical tools, and current knowledge.
Using cell line or animal model of BMBC, IL-6, TGF-β, and TFF genes were over-expressed.[3040414243] The recent article presented that 15 genes (APOPEC3B, ATL2, BBS1, C6orf61, C6orf167, MMS22L, KCNS1, MFAP3L, NIP7, NUP155, PALM2, PH-4, PGD5, SFT2D2, and STEAP3) were associated with the development of BMBC among patients.[43] Among 3 niches associated with BM, we considered the endosteal niche related with osteoblast as the critical key.[18] Therefore, we evaluated the causal network with the genes present in both BMBC and osteoblast, but excluded all of the genes present in the primary BC data set. Because of that, most signature genes, which reported in previous studies with BMBC, were not included in our study. We found 3 plausible upstream genes (NFKB2, UBIAD1, HEBP1) and 12 plausible downstream genes that had direct connection to BMBC. These 15 genes may be a good starting point to better understand the pathophysiology of BMBC. In turn, this will enable us to make efficient diagnosis and effective treatments of BMBC for patients who have primary BC without BM. Also, the plausible upstream genes could be powerful candidates for target therapy of BMBC. NFKB2, one of the plausible upstream genes, encode NF-κB protein that is a transcription factor known as a regulator of the immune system and promotes epithelial-to-mesenchymal transition and the metastasis of BC.[304445] Although the protein encoded by UBIAD1 gene was reported as bladder tumor suppressor protein, there was no report about its role in tumor development or the progression of HEBP1.[46] The UBIAD1 gene had 3 direct cause (parent) genes in S◆, i.e., DVL1, INPP1, and MAPK3. DVL1 and INPP1 are registered as BCRGs in KEGG (Fig. 6). In addition, UBIAD1 gene is one of the genes that are reported in minimum combination of gene expression patterns that show maximum RR in this study. Therefore, although a study for the direct effect of HEBP1 gene on BMBC will be needed, however, it is plausible that the UBIAD1 may have an effect on BMBC through influences of other genes such as DVL1 and MAPK3.
We mentioned some genes related with chemotherapy resistance and cancer progression in the Results section. The CBN models that we learnt in this study can explain how the genes interact to develop BMBC (group variable that represent either a subject has osteoblast or BM), drug resistance, cancer progression and metastasis as published in earlier studies.[313233343536373839] The role of those genes can be investigated as diagnostic candidates of BMBC in a future study. FOLH1 and ZFP36L2 had the remarkable changes of status in gene expression according to a subject with BMBC or osteoblast. Previous studies presented that the variants of proteins encoded by FOLH1and ZFP36L2 genes were potential contributors of risk toward breast, pancreas and prostate cancers.[3947] In the CBN structures presented in this study, the significant changes of FLOH1 AND ZFP36L2 according to state of group (BM or osteoblast) may suggest that the genes can be considered as candidates of diagnostic biomarker in BMBC. There were several limitations in the present study. Although the CBN in this study provided meaningful genes for BMBC and showed plausible causal structure among genes through machine learning techniques, the sample size were limited of only 48 human subjects and lacking the control subjects with neither of osteoblast or BM. Therefore, several signature genes related BM as CXCL12 and tumor necrosis factor-related apoptosis-inducing ligand, presented in the previous study using GSE 14017 and GSE 14018, were not selected in the final structure of the present study.[48] Despite having obtained excellent results on the validation tests, we should increase the number of human samples to get a point estimate closer to the mean value of the population. In parallel, we plan a future study to obtain orders of variables with probabilistic score using a new variable order search code that is currently being tested in Dr. Yoo's lab. Datasets used in this study that were downloaded from microarray GEO public repository had several limitations that are worth mentioning for proper interpretation of the results reported in this study: (1) there were gender ambiguity of donor osteoblast cell and normal bone tissue (thirteen subjects) making the result limited in generalizing among the women; (2) microarray data does not provide genomic sequences, resulting in unknown genomic variation information of subjects. Also, because the physiological osteoblasts and the osteoblasts in metastatic microenvironment are different, the responses in cancer metastasis may not be similar. Therefore, we should use the biologic data of the osteoblasts in metastatic site in the future study. However, we believe that the CBN in this study give us several leads for animal follow up studies or other human studies. Also we are planning to collect different types of gene expression data, e.g., RNA-sequencing, for validation and extension of the current causal inference statistical model that we have developed.
The CBN composed with 16 variables(3 direct causes and 12 direct effects of BMBC [group variable]) and seems to be a reasonable causal inference network with a high prediction rate. The direct cause and effect genes of BMBC may be useful candidates for early diagnosis and target therapy of BMBC. Among those genes, ZFP36L2 may be a meaningful candidate for a predictor of BMBC. In addition, CBN analysis may provide us with a more comprehensive understanding of the pathophysiology of BM.









 XML Download
XML Download