

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Cystinuria (OMIM #220100) is an autosomal recessive genetic disorder. Cystinuria leads to defects in transepithelial transporters for dibasic amino acids, including cystine, ornithine, lysine, and arginine. Although the urine concentration of all dibasic amino acids is elevated in cystinuric patients, only cystine leads to stone disease as it is relatively insoluble at physiological pH (1234). More than 50% of patients with cystinuria suffer from stone formation throughout their lifetime, with variable onset ages as well as a high rate of recurrence of up to 60% (5). Furthermore, since stones are likely to be formed bilaterally in more than three-quarters of cystinuric patients (4), they are at high risk of renal dysfunction and consequent poor quality of life (6).
Cystinuria types I, II, and III are traditional subgroups divided according to the urinary phenotype in heterozygotes (7). While type I heterozygotes show a normal amino aciduria, non-type I (type II and III) heterozygotes show high and moderate hyperexcretion of cystine and dibasic amino acids, respectively. Patients with type III, a mixed type, inherit type I and non-type I alleles from either parent. This classification system is still in clinical use. However, the International Cystinuria Consortium developed a new classification system to reflect the genetic and functional characteristics of the disease after the discovery of 2 genes responsible for cystinuria. Type A cystinuria is due to biallelic SLC3A1 mutations, type B cystinuria is due to biallelic SLC7A9 mutations, and type AB cystinuria is due to single heterozygous mutations in both genes (8). SLC3A1 encodes a heavy subunit of the renal cystine transport system, rBAT, and was identified as the cause of type I cystinuria. SLC7A9 encodes b0,+ amino acid transporter (b0,+AT), a light subunit of the renal cystine transport system and responsible for type II cystinuria. To date, 163 disease-causing mutations in SLC3A1 and 118 disease-causing mutations in SLC7A9 have been listed in The Human Gene Mutation Database (HGMD® Professional 2016.2, https://portal.biobase-international.com/hgmd/pro/start.php).
There have been several clinical case reports and studies of Korean patients with cystinuria (691011121314). However, only 2 case reports published include genetic studies: one was that of a 13-year-old boy with a single heterozygous SLC7A9 mutation (c.517G>A, p.G173R) (11), and the other was of an 8-month-old girl with a homozygous SLC3A1 mutation (c.1820delT, p.L607fs) (10). Here, we report a genotype-phenotype study of 8 Korean pediatric patients with cystinuria.
MATERIALS AND METHODS
Patients
Eight patients diagnosed with cystine stones during the period between January 2003 and June 2016 in 2 hospitals (Seoul National University Children's Hospital, Seoul, Korea and Asan Medical Center Children's Hospital, Seoul, Korea) were recruited. Their clinical presentation, clinical courses and serial laboratory findings were evaluated retrospectively.
Mutational studies
Genomic DNA was extracted from nucleated cells in peripheral blood using a commercial kit (QIAamp DNA Blood Mini Kit; Qiagen, Hilden, Germany). All coding exons and flanking introns of the SLC3A1 and the SLC7A9 genes were amplified using polymerase chain reaction followed by direct sequencing (primer sequences are available upon request).
RESULTS
Two of the 8 patients were male and six were female. The median ages at onset and diagnosis were 1.5 years (range, 0.3–13.6 years) and 2.6 years (range, 0.7–16.7 years), respectively. They were followed up for a median period of 7.7 years (range, 3.4–14.0 years). The clinical features of the patients are summarized in Table 1. The presenting symptoms were gross hematuria (n = 3) or urinary tract infection (n = 3) in association with renal stones, while renal stones were detected incidentally in two patients. Family history of nephrolithiasis was absent in all patients, except for one (Patient 7), whose maternal uncle had a history of nephrolithiasis of unidentified composition. The initial renal function was normal (estimated glomerular filtration rate [eGRF calculated using the Schwartz formula] ≥ 90 mL/min/1.73 m2) in 2 patients, and the remaining 6 patients had various degrees of renal dysfunction. Seven patients were treated with tiopronin (thiola®) with or without potassium citrate (n = 6) and captopril (n = 1). The remaining patient (Patient 5) had a single heterozygous SLC3A1 mutation and was treated with oral sodium bicarbonate only. However, all patients suffered from recurrent attacks of symptomatic nephrolithiasis, which lead to urologic interventions. At the last follow-up, 3 patients had a mild-to-moderate degree of renal dysfunction.
Table 1
Clinical features of 8 patients with cystinuria

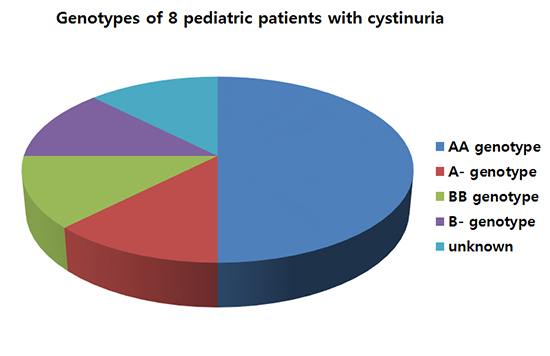
Urinary excretion of cystine and dibasic amino acids were markedly increased in all patients, except for Patient 5 who exhibited only a mildly increased level of cystine. The cyanide nitroprusside test was performed and positive in 2 patients (Table 2). Mutational analyses revealed biallelic SLC3A1 mutations (AA genotype) in 4 patients, a single heterozygous SLC3A1 mutation (A- genotype) in one patient, biallelic SLC7A9 mutations (BB genotype) in one patient, and a single heterozygous SLC7A9 mutation (B- genotype) in one patient. In one remaining patient, mutational analysis was not performed due to unavailable samples (Table 3). The c.1820delT SLC3A1 mutation was detected in 3 of 5 patients with an AA genotype (4 of 10 alleles).
Table 2
Urinary excretion levels (μM/g of creatinine) of branched amino acids of the patients
Table 3
Mutations detected in the patients

Urinary cystine excretion levels did not correlate with any clinical parameters or genotypes of the patients. We compared the clinical and laboratory findings of the 4 patients with the AA genotype to those of the others. The only difference observed was an older onset age in patients with the AA genotype. Urinary excretion levels of cystine did not correlate with any clinical parameters in the 4 patients with the AA genotype.
DISCUSSION
We report 8 cases of cystinuria, presenting with various symptoms. Mutational analyses revealed the AA genotype in 4 patients. Patients with the AA genotype had older onset ages than those with non-AA genotypes.
Several previous studies examining genotype-phenotype correlations in cystinuria did not show any correlation between patients with type A genotype and patients with non-A genotypes (81516). In a UK study (15), patients with at least one missense mutation in SLC3A1 had significantly lower levels of lysine, arginine, and ornithine, but not cystine, than patients with all other types of SLC3A1 mutations. Another UK study (16) revealed no difference between patients with type AA and patients with type BB in a variety of clinical parameters. In addition, patients with a single mutated allele also had variable disease severity and could not be differentiated from patients with 2 mutated alleles (16). In our study, the only difference between patients with the AA genotype and those with non-AA genotypes was the later age of onset in the former group. Of the 4 patients in our study with the AA genotype, Patient 4 had 2 truncating mutations and the youngest onset age. However, urinary excretion levels of cystine were highest in Patient 2, who had 2 missense mutations. Other clinical parameters, including long-term prognosis, showed no correlation with the different genotypes.
The SLC3A1 p.T216M mutation was detected in 2 of our patients. This mutation is common in South-eastern European (1718), Gypsy (19), and Greek populations (20) but has not been found in Chinese (21) and Japanese (22) patients. Conversely, the SLC7A9 p.P482L mutation is common in Japanese patients (22), but has not been found in European populations. In Patient 5, we found a nucleotide variation, c.1976A>C (p.Q659P), in SLC3A1. Using Mutation Taster (http://www.mutationtaster.org/), this variation was predicted to be a polymorphism, but was not found in ExAC (http://exac.broadinstitute.org/) or 1000G (http://www.1000genomes.org/). Mild but abnormal hyperexcretion of cystine and early onset nephrolithiasis in Patient 5 suggested that the p.Q659P variation may be a hypomorphic mutation. Patient 6 had a BB genotype. However, a study of the patient's family revealed type I cystinuria. A single heterozygous SLC7A9 mutation was detected in Patient 7. However, urinary excretion levels of branched amino acids in the patient were very high. Furthermore, her mother was heterozygous for the same mutation but was clinically silent, although urinary amino acid excretion levels were not measured in the mother. Therefore, it is quite possible that Patient 7 had another pathogenic mutation, not detected by Sanger sequencing, in SLC3A1 (AB genotype) or SLC7A9 (BB genotype). The genotype of Patient 8 was unavailable, but family study revealed type I cystinuria.
Progression to chronic kidney disease in patients with cystinuria has been reported to be between 5 and 17% (232425), and the prevalence of end stage renal disease has been reported to be up to 5% (26). In a recent UK study (16), the level of renal impairment observed was similar across all genotypes. Recently, a recent large retrospective study conducted by the French Cystinuria Group (27) showed that 5 (1.1%) of 442 patients with cystinuria progressed to end stage renal disease at a median age of 35.0 (11.8–70.7) years. Multivariate analyses revealed that progression to chronic kidney disease was associated with age, hypertension, severe damage of renal parenchymal defined as a history of partial or total nephrectomy, and so on (27). In our study, 3 of 8 patients had a mild degree of renal dysfunction at the last follow-up.
In conclusion, we have analyzed the genotypes and phenotypes of cystinuria in 8 pediatric patients and identified two novel mutations. We did not observe an association between clinical course and genotype, except for earlier onset age in patients with non-AA genotypes. A big limitation of this study was the small number of subjects. Therefore, a large nationwide multicenter study is recommended.
 XML Download
XML Download