

 PDF
PDF ePub
ePub Citation
Citation Print
Print



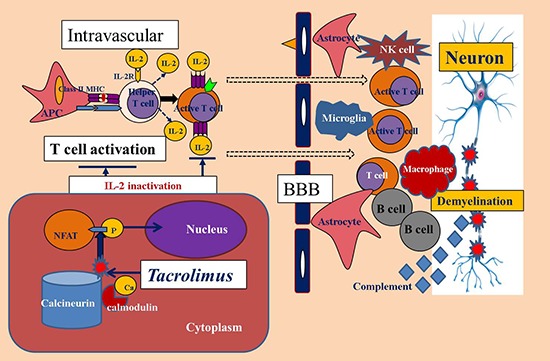
INTRODUCTION
Multiple sclerosis (MS) is a T-lymphocyte-mediated autoimmune disease that is characterized by relapsing and remitting inflammatory demyelination in the central nervous system (CNS) (123). Patients with MS exhibit long-term disabilities, such as weakness, sensory symptoms, decreased visual acuity, urination difficulties, and cognitive deficits (1234).
Previous studies on MS have concentrated on the mechanisms that are hypothesized to underlie the CNS inflammation, including T-cell autoimmunization, environmental factors, altered genetic influences, impaired vitamin D deficiency, and combinations of autoimmune diseases (1234). However, the underlying mechanisms and treatment of MS remain uncertain.
Recently, considerable progress has been made toward understanding the basic pathogenesis of MS and the effectiveness of disease-modifying therapies (DMTs) in preventing the relapse of patients with MS (5678). Although many DMTs are presumed effective in patients with MS, studies on the efficacy of DMTs for preventing the relapse of patients with MS are limited. In addition, DMTs can induce serious adverse effects (910).
We tested the anti-inflammatory effects of an oral-formulated immunosuppressant with a better side effect profile in a mouse model of experimental autoimmune encephalomyelitis (EAE) (11). Tacrolimus (FK506; macrolide lactone immunosuppressant) acts as a calcineurin inhibitor that blocks interleukin-2 production, which results in decreased T-cell proliferation (1213). Tacrolimus is therefore used in the treatment of T-cell-mediated autoimmune diseases and prevention of organ transplant rejection (1213). The EAE animal model is used most in studies of MS in laboratory animals. Both MS and EAE are characterized by perivascular inflammation and demyelination in the spinal cord and brain. EAE is a CD4+ T-cell-mediated autoimmune disease in which CNS inflammation is induced after the animals are immunized against a myelin-specific antigen that induces the migration of activated autoreactive T-cells across the blood-brain barrier and into the CNS (1415).
In this study, we used C57BL/6 mice that had been immunized against the myelin oligodendrocyte glycoprotein (MOG35–55) peptide, which is a method that is widely applied to induce EAE in animals. Because of the promising beneficial effects and safety of tacrolimus, the present study aimed to assess the therapeutic effects of oral tacrolimus in MS.
MATERIALS AND METHODS
Induction of EAE and animal care
All of the procedures were performed in accordance with the Institutional Animal Care and Use Committee (IACUC) guidelines for the care and use. Animals exhibiting paralysis were kept on soft bed of each cage and fed and watered through animal feeding tube. All surgery was performed under sodium pentobarbital anesthesia, and all efforts were made to minimize suffering. If any mouse came to the moribund stage, it was decapitated after anesthesia with sodium pentobarbital.
Sixty-four mice that were approximately 80 days old were randomly divided into 3 experimental groups: an untreated EAE group, a 5-mg/kg tacrolimus-treated EAE group, and a 10-mg/kg tacrolimus-treated EAE group. During the week before the start of and during the experiments, the animals were housed in individual cages with an ambient temperature of 23°C ± 3°C, stable air humidity, and a natural day-and-night cycle. The adoptive transfer of EAE was performed as described previously. Briefly, 11-week-old female C57BL/6 mice (Central Lab. Animal Inc., Seoul, Korea) were immunized against the MOG35-55 (MEVGWYRSPFSRVVHLYRNGK) peptide in incomplete Freund's adjuvant supplemented with Mycobacterium tuberculosis (EAE induction Hooke kits™, Catalog No. EK-2110; Hooke Laboratories, Inc., Lawrence, MA, USA) (161718). Each mouse was immunized subcutaneously with MOG (200 μg), and pertussis toxin (200 ng) was intraperitoneally injected once. The animals were sacrificed 14 days after the autoimmunization (1920). During the 14 days before the sacrifice of the EAE mice, regular doses (5 or 10 mg/kg) of oral tacrolimus (Prograf®; Astellas Pharma Inc., Tokyo, Japan) were administered orally once per day beginning after the autoimmunization of the tacrolimus-treated EAE groups. The untreated EAE group was used for comparison.
Clinical characteristics of EAE
All of the animals were checked daily after tacrolimus treatment, and their symptoms were recorded. EAE mice show flaccid paralysis, which is characterized by decreased muscle tone that progresses from the tail upward through the body (1619). A 6-point scale (0–5) was used to rate the severity of the symptoms, with a score of 1 denoting tail paralysis, a score of 4 indicating quadriplegia, and a score of 5 signifying death. An investigator who was blind to the groups rated the experimental animals as follows: 0, no clinical disease; 0.5, piloerection; 1, tail weakness; 1.5, tail paralysis; 2, hind limb weakness; 3, hind limb paralysis; 3.5, forelimb weakness; 4, forelimb paralysis; or 5, moribund or death. The symptom scores of the mice are expressed in mean ± standard error. The data were analyzed with analysis of variance tests and Dunnett's post-hoc tests, if necessary (SPSS, version 19.0; IBM Corp., Armonk, NY, USA). P values less than 0.05 were considered statistically significant.
Immunoblotting and antibodies
Fourteen days after immunization, each mouse was perfused with phosphate-buffered saline (PBS) while they were deeply anesthetized, and the CNS was removed. The spinal cords were homogenized in ice-cold radio immunoprecipitation assay buffer containing protease inhibitor and phosphatase inhibitor cocktails (Catalog No. 01906845001 and 11697498001; Roche Diagnostics Corporation, Indianapolis, IN, USA). The samples were centrifuged at 14,000 rpm for 25 minutes at 4°C, and the supernatants were collected. The protein concentrations of the samples were measured with Thermo Scientific Pierce Micro BCA Protein Assay Kits (Thermo Fisher Scientific Inc., Waltham, MA, USA). The samples were denatured in loading buffer for 10 minutes at 95°C, loaded into 15% gel for separation by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and then blotted onto nitrocellulose membranes with a Trans-Blot Cell system (Bio-Rad Laboratories, Inc., Hercules, CA, USA). After the transfer, the membranes were blocked for 1 hour at room temperature with 5% skim milk (Duchefa Biochemie B.V., Haarlem, The Netherlands) in Tris-buffered saline (20 mM Tris, 150 mM NaCl [pH 7.5]) with 0.1% Tween 20 (TBST). The membranes were then incubated overnight at 4°C with one of the following primary antibodies: MOG (1:1,000, Catalog No. ab109746; Abcam plc, Cambridge, UK), myelin basic protein (1:1,000, Catalog No. ab40390; Abcam plc), glial fibrillary acidic protein (GFAP, 1:4,000, Catalog No. ab7260; Abcam plc), ionized calcium binding adaptor 1 (Iba1, 1:250, Catalog No. ab48004; Abcam plc), or CD4 (1:200, Catalog No. sc-1140; Santa Cruz Biotechnology, Inc., Dallas, TX, USA). The membranes were then washed in TBST buffer and incubated for 1 hour with a horseradish peroxidase (HRP)-conjugated goat anti-rabbit immunoglobulin G (IgG) antibody (1:5,000, Catalog No. ADI-SAB-300; Enzo Life Sciences, Inc., Farmingdale, NY, USA) or HRP-conjugated donkey anti-goat IgG (1:2,000, Catalog No. A50-101P; Bethyl Laboratories, Inc., Montgomery, TX, USA) that was diluted in 5% (w/v) skim milk in TBST buffer. The membranes were then washed in TBST and developed with a Chemiluminescent Sensitive Plus HRP Microwell and/or Membrane Substrate (Surmodics, Inc., Eden Prairie, MN, USA). The membranes were stripped with Restore Western Blot Stripping Buffer (Thermo Fisher Scientific Inc.) at room temperature for 20 minutes and then incubated with an antibody to β-actin (1:1,000, Catalog No. 4970; Cell Signaling Technology, Inc., Danvers, MA, USA), which was used as a loading control.
Immunohistochemistry
After the mice were killed and the spinal cord removed, the spinal cord was fixed with 4% (w/v) paraformaldehyde in PBS buffer overnight at 4°C. The spinal cords were dehydrated in 30% sucrose at 4°C until the tissue sank. The tissue was then embedded in O.C.T. compound (Sakura Finetek USA, Inc., Torrance, CA, USA) and frozen with dry ice (2122). The samples were cut into 10-μm coronal sections from the cervical spine to thoracic spine. The sections were stained with Luxol fast blue (LFB) stain or hematoxylin & eosin (22). For the LFB stain, which was performed to stain myelin, the sections were incubated overnight at 60°C in 0.1% LFB (Solvent Blue 38; Sigma-Aldrich Co., LLC, St. Louis, MO, USA). The sections were rinsed in distilled water (DW) twice and then differentiated by dipping them in 0.01% lithium carbonate (Cat#L0224; Tokyo Chemical Industry Co., Ltd., Tokyo, Japan). The sections were rinse twice in DW and then Nissl stained by incubating them in 0.1% cresyl violet (Sigma-Aldrich Co., LLC) for 5 minutes (2223). The sections were then rinsed quickly in fresh DW. The sections were dehydrated by dipping them in 3 changes of absolute ethanol (Merck Chemicals GmbH, Darmstadt, Germany). For the hematoxylin & eosin, which was performed to analyze inflammation, the tissue sections were hydrated in DW for 10 minutes, stained with Mayer's Hematoxylin solution (Sigma-Aldrich Co., LLC) for 10 minutes, washed in DW for 5 minutes, and then stained with Eosin Y solution, alcoholic (Sigma-Aldrich Co., LLC) for 3 minutes (2223). The sections were rinsed quickly in 70% Ethanol and dehydrated in 3 changes of absolute ethanol (Merck Chemicals GmbH). All of the sections were mounted on slides and cover slipped with Permount (Thermo Fisher Scientific Inc.). Light microscopy was used to examine the slides.
RESULTS
In this study, we evaluated the clinical EAE symptom scores in each EAE group every day during the 14-day study in order to examine the functional loss and alterations of the mice in this EAE model. In addition, we examined inflammatory cell infiltration and demyelination in the spinal cord specimens with immunohistochemistry to determine if the severity of the spinal cord histopathology correlated with tacrolimus administration. Furthermore, we investigated autoimmunization and CNS inflammation by evaluating the presence of activated microglial cells, interleukin-2, macrophages, and T-cell activation in the EAE animals.
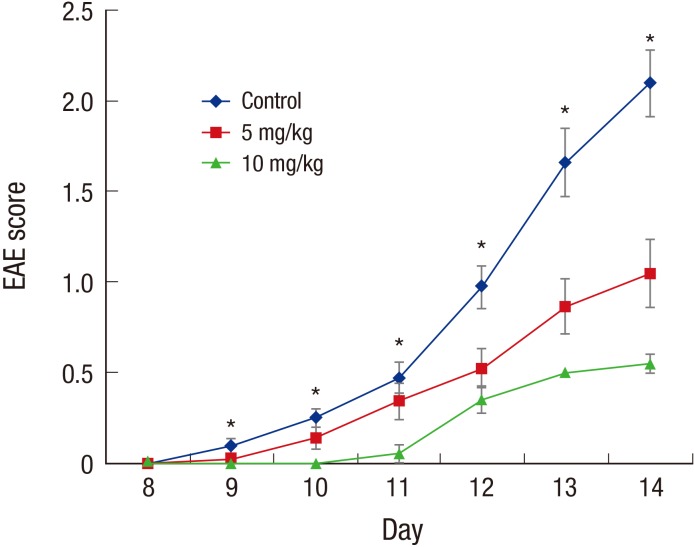
After autoimmunization with MOG35–55, the EAE symptom scores increased gradually in each group over time, and significant differences were found among the groups beginning the 10th day after immunization (Fig. 1; P < 0.05). The tacrolimus-treated mice had lower symptom scores compared with the untreated mice. The 10-mg/kg tacrolimus-treated group had lower EAE symptom scores compared with the 5-mg/kg tacrolimus-treated group. These results demonstrated that the oral form of tacrolimus improved the clinical symptoms and disease progress of the EAE mice model.
Fig. 1
Behavioral tests of EAE mice. After autoimmunization with MOG35–55, the symptom scores of each EAE group gradually increased over time, and significant differences were found among the 3 groups (P < 0.05) beginning on the 10th day. The 10-mg/kg tacrolimus-treated EAE mice exhibited lower EAE scores, which suggested that they had less clinical symptoms, compared with the untreated EAE mice and 5-mg/kg tacrolimus-treated EAE mice.
EAE = experimental autoimmune encephalomyelitis, MOG = myelin oligodendrocyte glycoprotein.
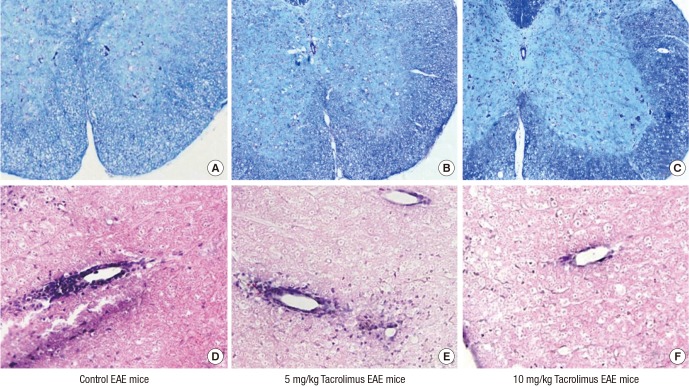
We stained the spinal cord sections with LFB to visualize myelin and the demyelination zones in the white matter (Fig. 2). Tacrolimus treatment remarkably maintained the levels of myelination in the spinal cords of the EAE mice at levels similar to those in control mice. The untreated EAE mice showed marked demyelination in the white matter of the spinal cord and inflammatory cell infiltration in the perivascular area. The tacrolimus-treated EAE groups exhibited higher myelination levels and decreased inflammation compared with the control EAE mice. In addition, compared with the 5-mg/kg tacrolimus-treated EAE mice, the 10-mg/kg tacrolimus-treated EAE mice had higher levels of myelination and less inflammatory infiltration, and cuffed vessels were observed in the spinal cord. Tacrolimus treatment markedly inhibited autoimmunization and the infiltration of inflammatory cells into the spinal cords of the EAE mice. These results demonstrated that tacrolimus treatment inhibited the immune cascade abnormal infiltration of inflammatory cells, and spinal cord demyelination of the EAE mice.
Fig. 2
Histopathology of EAE mice. The spinal cord specimens of the untreated EAE mice exhibited marked demyelination (A) and inflammatory cell infiltration in the perivascular area (D). However, the tacrolimus-treated EAE mice exhibited preserved myelination and decreased inflammation compared with the untreated group. Additionally, the 10-mg/kg tacrolimus-treated EAE mice exhibited more myelination (C) and decreased inflammation (F) compared with the 5-mg/kg tacrolimus-treated EAE mice (B, E).
EAE = experimental autoimmune encephalomyelitis.
In order to determine the clinical significance of the tacrolimus-mediated decreases in CD4 T-cell activity, we investigated whether tacrolimus prevented the clinical symptoms of EAE in the mice. During tacrolimus treatment, the in vivo functions of the CD4 cells were blocked in the EAE mice. Taken together, these results suggested that CD4 T-cell activity plays an important role in the tacrolimus-mediated decrease in inflammatory reaction by autoimmunity in EAE model.
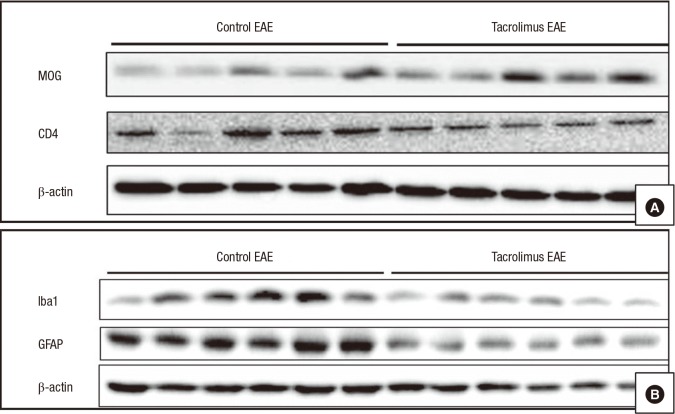
Western blotting was used to compare the levels of immunization biomarkers in the untreated and tacrolimus-treated EAE groups (Fig. 3). Compared with the untreated EAE mice, the tacrolimus-treated EAE mice exhibited stronger immunoreactivity to MOG and weaker immunoreactivity to CD4, Iba1, and GFAP. Collectively, these results suggested that tacrolimus suppressed the EAE in the mice by inhibiting T-cell activation.
Fig. 3
Western blots of EAE mice. Fourteen days after the immunization, specimens from the untreated EAE group and tacrolimus-treated groups were investigated with western blots. The blots of the samples from the tacrolimus-treated EAE mice contained bands with strong immunoreactivity to MOG and weak immunoreactivity to CD4 compared with those in the untreated mice (A). In addition, the blots of the samples from the tacrolimus-treated EAE mice had weak immunoreactivity to Iba1 and GFAP compared with those in the untreated EAE mice (B).
EAE = experimental autoimmune encephalomyelitis, MOG = myelin oligodendrocyte glycoprotein, Iba1 = ionized calcium binding adaptor 1, GFAP = glial fibrillary acidic protein.
DISCUSSION
In this study, we evaluated the anti-inflammatory effects of tacrolimus in an EAE mouse model and found that tacrolimus decreased the severity of the clinical symptoms of EAE and inhibited autoimmunization and inflammation in the EAE model. Thus, these results suggested that tacrolimus might be effective in the treatment of MS. EAE in mice and MS are thought to have similar causes: the infiltration of autoreactive T-cells and associated inflammatory cells into the CNS, which results in the immune-associated CNS demyelination that is observed in patients with MS and EAE animals (2425).
MS is a devastating demyelinating disease of the human CNS, and expensive DMTs are currently used to treat patients with MS (26). However, therapies that effectively prevent the relapse of MS are not yet available. In particular, the limited efficacy and severe side effects, as well as treatment costs, of these therapies often limit their availability. The side effects include flu-like symptoms, menstrual disorders in women, decreased neutrophil and white blood cell counts, progressive multifocal encephalopathy, cardiac problems, increased aspartate transaminase and alanine transaminase levels, and the development of neutralizing antibodies (9102728). Therefore, it is necessary to investigate safe, effective, and less expensive therapeutic options for patients with MS.
The results of the present study suggested that tacrolimus might be useful for treating patients with MS because of its neuroprotective properties, safety, and reasonable cost. The oral form of tacrolimus, which has been clinically approved for other diseases (1213), has recently been shown to have significant immunosuppressive properties that result in the inhibition of CNS demyelination and axonal injury in EAE mice and patients with MS. Furthermore, these results suggest that the anti-inflammatory properties of immunosuppression are responsible for the decreased clinical symptoms in the EAE mice, and these properties might be critical for the effective inhibition of relapse in patients with MS.
Relatively few studies have been conducted on the use of tacrolimus as a treatment for immune-associated CNS diseases, and, to the best of our knowledge, no studies have investigated the use of the approved oral form of tacrolimus in EAE model. Of course, a few studies revealed therapeutic pathomechanisms of tacrolimus on EAE model by the peritoneal injections of FK506, however our study identified therapeutic effectiveness of oral formulated tacrolimus which would be much feasible as strategic therapeutics compared with injection (2930). Also, a clinical study on the combination therapy of tacrolimus and interferon beta was reported, however it did not identify pure therapeutic effects of oral formulated tacrolimus in MS because interferon therapy has been established DMT for MS (31).
However, the results of our study provide evidence that the oral administration of tacrolimus to mice suppressed the disease process underlying EAE, inhibited the invasion of mononuclear cells into the spinal cord, and restored myelination in the CNS. Additionally, we did not observe any side effects in any of the mice treated with tacrolimus. Therefore, this study would be the cornerstone for further clinical researches on MS with using tacrolimus.
In summary, these results revealed that tacrolimus was therapeutic by inhibiting autoimmunization in EAE mice. These therapeutic effects of tacrolimus might result in the inactivation of the CD4 T-cell immune pathway and decreased inflammatory cells. In conclusion, the results of the present study suggested that the oral administration of tacrolimus might be an ideal alternative DMT for patients with MS because of its safety, anti-inflammatory effects, and affordability. Further studies are required to identify the therapeutic dose of tacrolimus in patients with MS.
 XML Download
XML Download