

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
The global influenza surveillance network established by the World Health Organization (WHO) in 1948 has allowed for the prediction of the specific influenza virus strains to be targeted by the following flu season's influenza vaccines. Supported by this effort, the first trivalent influenza vaccine was formulated, containing antigens for 2 subtypes of the influenza A virus (H1, H3) and 1 influenza B virus.1 Influenza B viruses, which cause symptoms similar to those of influenza A viruses, can lead to death in severe cases. Influenza B viruses are also known to be associated with Reye syndrome. Although influenza B viruses are not divided into subtypes, they can be further classified into 2 antigenically distinct lineages known as the Victoria and Yamagata lineages. It has been observed that 1 of these lineages tends to become predominant during each flu season. Each year, after a review of worldwide surveillance data, the WHO recommends a composition for the influenza vaccines to be manufactured for the upcoming flu season. Unfortunately, in the case of a mismatch between the recommended vaccine lineage and the predominantly circulating lineage, little cross-immunogenicity is expected to occur. In fact, mismatches between the WHO recommended virus strains and the circulating virus strains have occurred in 6 of the past 11 years.
Of the influenza viruses classified over the past 11 years, up to 46% have been influenza B viruses. Since 2001, the frequency with which the 2 B lineages have been found to circulate concurrently in a single season has been on the rise, a trend which has spurred the need for a quadrivalent influenza vaccine formulated to protect against both B lineages. Reflecting this reality, the WHO recommended that trivalent or quadrivalent flu vaccines include one or both B lineages beginning in the 2013–2014 flu season.2 A recent study also reported that quadrivalent influenza vaccines can reduce the risks of flu-associated hospitalization and mortality relative to that of trivalent influenza vaccines.3
Since October 2008, Green Cross Corporation (Yongin & Hwasun, Korea) has produced influenza vaccines that met the recommendations of the European Pharmacopoeia and the WHO. Since 2009, the pharmaceutical company has been selling GC Flu (a split-virion, inactivated trivalent influenza vaccine) as well as pre-filled syringes of GC Flu. The study vaccine, GC3110A, is a fertilized egg-based, split-virion, quadrivalent influenza vaccine containing antigens for both B virus lineages. Results of a phase 1/2a clinical trial indicated that GC3110A elicited immunogenic responses against 4 influenza virus strains, A/H1N1, A/H3N2, B/Yamagata, and B/Victoria. A phase 3 clinical trial involving healthy adults also found that GC3110A was non-inferior to the control trivalent influenza vaccines (GC Flu pre-filled syringes) in terms of its immunogenicity against all flu vaccine virus strains. This clinical trial also found that GC3110A did not result in any increased risk of particular adverse events compared with those of the control vaccines, demonstrating that this new influenza vaccine is effective and safe. Following up on the phase 1/2a trial and phase 3 trial in healthy adults, the present clinical trial aimed to evaluate the efficacy (immunogenicity) and safety of the quadrivalent, inactivated, split-virion influenza vaccine (GC3110A) in healthy Korean children and adolescents aged ≥ 6 months to < 19 years.
METHODS
Study design
The purpose of the present clinical trial was to evaluate the efficacy (immunogenicity) and safety of a quadrivalent, inactivated, split-virion influenza vaccine (GC3110A) in healthy children and young adults aged ≥ 6 months to < 19 years. The trial consisted of 2 parts, each designed to evaluate the safety and immunogenicity of the vaccine being tested. Part 1 was planned as a single institution, open-label, and single group clinical trial, and part 2 was a multi-institutional, double-blinded, randomized, active-controlled clinical trial.
Part 1
All prospective subjects volunteering to participate in the trial and their legal guardians were required to give written informed consent prior to trial entry (legal guardian consent only was required for prospective subjects under the age of 7 years). Following the consent process, the necessary physical examinations and testing detailed in the protocol were administered to the prospective subjects. Those who met the entry criteria were then assigned to experimental groups and received one or two doses of the vaccine according to their age.
The Data Safety Monitoring Board (DSMB) reviewed all the solicited adverse events recorded during the 7-day period following the administration of each vaccine in part 1. However, in the absence of adverse events assessed at or above grade 3 as defined by the Food Drug Administration (FDA)'s Toxicity Grading Scale for Healthy Adults and Adolescents,4 the trial proceeded to part 2 without DSMB review.
Randomization was not used in part 1 of the study, which was a 15-subject, single group clinical trial designed to evaluate the safety of the study vaccine. Subsequent procedures and methods used to evaluate the vaccine's immunogenicity and safety, as well as visitation schedule, were the same as those used in part 2.
Part 2
All prospective subjects volunteering to participate in the trial and their legal guardians were required to give written informed consent prior to trial entry (legal guardian consent only was required for prospective subjects under the age of 7 years). Following the consent process, the necessary physical examinations and testing detailed in the protocol were administered to the prospective subjects. Those who met the trial entry criteria were then randomized in a 4:1 ratio into the experiment group or the control group to receive one or two doses of vaccine according to their age.
Subjects aged ≥ 6 months to < 3 years received a 0.25 mL dose of vaccine, while subjects aged ≥ 3 years to < 19 years received a 0.5 mL dose of vaccine. Subjects younger than 9 years of age who had not been previously been immunized against influenza received a second dose of vaccine 28 days following the first dose.
Vaccines
The study vaccine, GC3110A, a quadrivalent split-virion influenza vaccine developed by Green Cross Corporation, is an inactivated influenza vaccine derived from fertilized eggs. Each 0.5 mL dose of the vaccine contained 15 µg of each of the four purified hemagglutinin antigens (60 µg total): A/California/7/2009 (H1N1), A/Switzerland/9715293/2013 (H3N2), B/Phuket/3073/2013, and B/Brisbane/60/2008.
The control vaccine, a GC Flu pre-filled syringe containing a trivalent split-virion influenza vaccine, was also developed by Green Cross Corporation. It is an inactivated influenza vaccine derived from fertilized eggs. A 0.5 mL dose of the vaccine contained 15 µg of each of the three purified hemagglutinin antigens (45 µg total): A/California/7/2009 (H1N1), A/Switzerland/9715293/2013 (H3N2), and B/Phuket/3073/2013.
Subjects
The present trial included healthy children and young adults aged ≥ 6 months to < 19 years. Exclusion criteria included a history of hypersensitivity to any component of the vaccines, including eggs and chicken meat; receipt of an influenza vaccine during the previous 6 months; immunosuppressive disorder including immune deficiency; history of Guillain-Barré syndrome; Down syndrome or cytogenetic disorder; moderate to severe chronic illness potentially interfering with full participation in the trial as determined by the researcher; any coagulation disorder contraindicating intramuscular injection or receipt of coagulating agent; acute febrile illness (temperature ≥ 38.0°C) within 72 hours prior to scheduled receipt of vaccine in the trial; receipt of vaccine (of any kind) within 28 days prior to screening; receipt of immunosuppressive or immunomodulating agents within 3 months prior to screening; receipt of immunoglobulin or blood derived products within 3 months prior to screening; anticipated receipt of immunoglobulin or blood derived products during the trial period; fever/pain reliever (analgesic) or non-steroidal anti-inflammatory drug within 4 hours prior to scheduled receipt of vaccine in the trial; participation in a clinical trial within 28 days prior to screening; and other significant medical or psychiatric conditions deemed inappropriate for trial entry by the researcher.
Safety assessment
Adverse events reported by the subjects or identified in interviews were recorded according to the trial schedule. In step 1, adverse events experienced during the 28-day period following vaccination were surveyed. In step 2, serious adverse events experienced during the 180-day period following the final dose in step 1 were surveyed.
At the first visit, parents and legal guardians were provided with a thermometer and a diary card listing solicited local and generalized adverse events. They were also given a brief training regarding the use of the card for the daily recording of any solicited adverse events experienced during the 7-day period following vaccination and of any unsolicited adverse events experienced during the 28-day period following vaccination. At the follow-up visit, serious adverse events experienced during the 180-day period following vaccination were recorded, and their associations with the vaccines used in the trial were evaluated. Any serious adverse events were reported within 24 hours of detection by the researcher.
Immunogenicity assessment
Blood sampling for immunogenicity assays was performed at the first visit (before vaccination) and at the final visit (28 days after the final dose). Analysis was performed at the Vaccine and Bio Research Center of the Catholic University of Korea.
The primary efficacy (immunogenicity) end points following vaccination were the proportion of subjects who achieved seroconversion, defined as a pre-vaccination (day 0) hemagglutination inhibition (HI) antibody titer of less than 1:10 with a post-vaccination titer of > 1:40 at day 28 (case 1) or a pre-vaccination (day 0) titer of > 1:10 with a ≥ 4-fold increase in antibody titer at day 28 (case 2), and the proportion of subjects who achieved seroprotection, defined as an HI antibody titer of > 1:40 at day 28 following vaccination. The secondary efficacy (immunogenicity) end points following vaccination were the geometric mean titer (GMT) and GMT ratio (GMR), as measured with pre-vaccination (day 0) and post-vaccination (day 28) HI antibody titers.
Statistical analysis
The primary objective of the present trial was to determine whether the seroconversion and seroprotection rates (SPRs) on day 28 following the final dose of GC3110A met the FDA's approval standards for an inactivated seasonal influenza vaccine.5 To this end, it must be demonstrated that the lower boundary of the 95% confidence interval (CI) for the percentage of subjects achieving seroconversion for HI antibody meets or exceeds 40% and that the lower boundary of the 95% CI for the percentage of subjects achieving seroprotection for HI antibody meets or exceeds 70%. To demonstrate this, the proportion of subjects who achieved seroconversion for HI antibody and the proportion of subjects who achieved seroprotection for HI antibody were identified, and the two-sided 95% CIs for each strain were determined. The two-sided CIs for seroconversion and seroprotection were estimated based on the Clopper-Pearson interval.
To evaluate the safety of the study vaccine across various age groups, a total of 15 subjects (5 from each age group) were enrolled in part 1 of the clinical trial. Although the safety and tolerability of GC3110A in adults was verified in a phase 1/2a clinical trial, because the present clinical trial is the first to administer the vaccine in children and young adults, its safety among this population group was to be tested on the smallest possible sample prior to proceeding to part 2.
The rationale for tabulating the sample size in part 2 was as follows. In part 2, the testing power of each hypothesis for each strain was set at 97.25% to obtain an overall power of 80%. Here, the minimum expected SPR is 70%. Based on the combined results of a phase 3 clinical trial (GC501_C_P3)6 and a GC Flu pre-filled syringe phase 4 clinical trial (GCFLU_P4),7 the study vaccine's expected SPR for strain B Yamagata in healthy infants and young children was 80.3%. The minimum expected seroconversion rate (SCR) was 40%. Based on the combined results of a phase 3 pediatric clinical trial (GC501_C_P3) and phase 4 GC Flu pre-filled syringe phase 4 clinical trial (GCFLU_P4), the study vaccine's expected SCR for strain B in healthy infants and children was 51.3%.
Assuming the above SCRs, 295 subjects would be needed without accounting for attrition; with an attrition rate of 30%, 422 subjects would be needed. Based on a 4:1 ratio between the experiment group and the control group, 106 subjects would be required in the control group. Thus, allowing for a 30% attrition rate, a total of 528 subjects were recruited (experiment: 422, control: 106).
For immunogenicity and safety analysis, the data collected through step 1 were utilized. Incorporating the data on serious adverse events obtained through step 2, additional safety analysis was performed. The results of the safety analyses in parts 1 and 2 were combined for presentation. Missing values were not substituted, and all tests were two-sided with a significance level (α) of 5%, unless otherwise stated. Descriptive statistics are presented for all continuous data (sample size, mean, standard deviation, minimum, median, maximum), while frequency and percentage are presented for all categorical data. If needed, two-sided 95% CIs are presented. All statistical analyses were performed using SAS version 9.4 (SAS Inc., Cary, NC, USA).
Ethics statement
The present study was conducted in accordance with the Korean Good Clinical Practice (KGCP) standards of the Korean Ministry of Food and Drug Safety (MFDS) and the International Conference on Harmonisation (ICH) for all steps involved, including protocol design, maintenance of records, collection of data, and obtaining of Institutional Review Board (IRB) approval. This is a randomized clinical trial on the third phase, registered at the ClinicalTrials.gov (https://www.clinicaltrials.gov/) number NCT02541253.
Throughout its duration, the study adhered to the principles of the Declaration of Helsinki, which prioritize the rights and safety of subjects participating in clinical trials. In accordance to the current clinical trial protocol approval process, MFDS approval (Clinical Trial Management Division 3114) was obtained on June 25, 2015 prior to proceeding with the present trial. Part 1 of the trial was approved as a single institution study conducted at the Catholic University of Korea's Seoul St. Mary's Hospital, while part 2 was approved as a multi-institutional study conducted across a total of 12 institutes. Prior to the beginning of the trial, the clinical trial protocol and all other associated items were submitted to and approved by each relevant IRB beginning with the St. Mary's Hospital IRB approval on June 17, 2015 (IRB No. KC15BDMT0412).
RESULTS
Study subjects
For part 1, a total of 15 subjects were recruited between September 12, 2015 and March 17, 2016. For part 2, a total of 528 subjects were recruited between October 12, 2015 and June 27, 2016. All 15 subjects in part 1 met the screening criteria and were assigned to the experiment group. Upon conclusion of the clinical trial, they were included in the safety analysis group. The 528 subjects enrolled in part 2 were randomized in a 4:1 ratio into the experiment and control groups, resulting in 422 subjects in the GC3110A group and 106 subjects in the GC Flu pre-filled syringe group. After excluding 1 subject in the GC3110A group who withdrew their consent, a total of 421 subjects received the study vaccine and went on to complete the trial (Fig. 1). The subjects' demographic characteristics are presented in Table 1. Basic demographic variables of the subjects, including age, age group distribution, height, body weight, medical history, and medication history, were similar between the GC3110A group and the GC Flu pre-filled syringe group.

Table 1
Demographic characteristics of the subjects

![]()
Immunogenicity
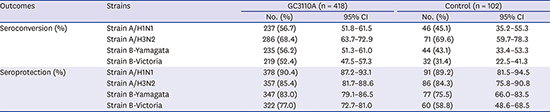
Per-protocol analysis revealed that the proportion of subjects in the GC3110A group who achieved post-vaccination seroconversion based on the HI antibody titer against each vaccine strain was 56.7% for A/H1N1, 68.4% for A/H3N2, 56.2% for B/Yamagata, and 52.4% for B/Victoria. The lower limits of the two-sided 95% CIs for the SCRs of the subtypes were 51.8%, 63.7%, 51.3%, and 47.5%, respectively, all of which exceeded the recommended 40%. In the control group (GC Flu pre-filled syringe), the proportion of subjects who achieved seroconversion was 45.1% for A/H1N1, 69.6% for A/H3N2, 43.1% for B/Yamagata, and 31.4% for B/Victoria.
Per-protocol analysis also revealed that the proportion of subjects in the GC3110A group who achieved post-vaccination seroprotection based on HI antibody titer against each vaccine strain was 90.4% for A/H1N1, 85.4% for A/H3N2, 83.0% for B/Yamagata, and 77.0% for B/Victoria. The lower limits of the two-sided 95% CIs for the SPRs of the subtypes were 87.2%, 81.7%, 79.1%, and 72.7%, respectively, all of which exceeded the recommended 70%. In the control group (GC Flu pre-filled syringe), the proportion of subjects who achieved seroprotection was 89.2% for A/H1N1, 84.3% for A/H3N2, 75.5% for B/Yamagata, and 58.8% for B/Victoria (Table 2).
Table 2
Immunogenicity endpoint results

![]()
For subanalysis of the vaccine's immunogenicity by age group, subjects were classified into the following age groups: ≥ 6 months to < 3 years, ≥ 3 years to < 9 years, ≥ 9 years to < 19 years. We confirmed that the proportion of subjects who achieved seroconversion for all four vaccine strains exceeded 40% in the GC3110A group (Table 3). In subjects aged ≥ 6 months to < 3 years, the proportion in the GC3110A group who achieved post-vaccination seroprotection based on HI antibody titer against each vaccine strain was 82.9% for A/H1N1, 68.5% for A/H3N2, 63.1% for B/Yamagata, and 49.5% for B/Victoria. In all other age groups, SPRs for all four vaccine strains exceeded 70% (Table 4).
Table 3
Percentage of subjects with seroconversion by influenza strain and age group

![]()
Table 4
Percentage of subjects with seroprotective levels of antibodies by influenza strain and age group

![]()
Safety
The rates of total adverse events, regardless of any causal relationship with the vaccines used in the study, were 67.6% in the GC3110A group (296/438 subjects, 944 cases) and 56.7% (59/104 subjects, 166 cases) in the GC Flu pre-filled syringe group. The solicited localized adverse events and solicited generalized adverse events that occurred in step 1 of part 1 and part 2 are summarized and presented in Table 5. Of the reported localized adverse events, pain and tenderness were the most common in both the GC3110A group and the GC Flu pre-filled syringe group. A total of 47.0% of subjects in the GC3110A group reported tenderness (206 subjects, 215 cases), and 39.4% of subjects in the control group reported pain (197 subjects, 206 cases). In terms of solicited generalized adverse events, fatigue and malaise were the most common in both the GC3110A and control groups.
Table 5
Solicited adverse events

![]()
The rates of unsolicited adverse events during the 28-day period following vaccination were 24.0% (105/438 subjects, 168 cases) in the GC3110A group and 20.2% in the control group (21/104 subjects, 33 cases). In the GC3110A group, nasopharyngitis (7.5%) and upper respiratory tract infections (4.6%) were the most common. Likewise, nasopharyngitis (8.7%) and upper respiratory tract infections (6.7%) were the most common unsolicited adverse events in the control group.
All subjects were observed for 30 minutes following vaccination for signs of immediate adverse reactions to the vaccines. However, no such cases were reported. A total of 7 serious post-vaccination adverse events were reported during step 1 of parts 1 and 2, resulting in rates of 0.9% (4/438 subjects, 6 cases) in the GC3110A group and 1.0% (1/104 subjects, 1 case) in the control group. All reported serious adverse events, which were classified as unsolicited adverse events, were determined to be unrelated to the vaccines used in the trial. Subjects who participated in parts 1 and 2 were tracked and assessed 180 days following the final dose (step 2). Including the serious adverse events observed during step 1 of parts 1 and 2, a total of 31 adverse events were observed, including 20 cases in the GC3110A group (3.9%, 17/438 subjects) and 11 cases in the control group (4.9%, 5/103 subjects). All reported serious adverse events were determined to be unrelated to the vaccines used in the study.
DISCUSSION
The present clinical trial is the first open-label, single group (part 1), randomized, double-blinded, and activate-controlled phase 3 clinical trial to evaluate the efficacy and safety of a novel quadrivalent split-virion influenza vaccine in healthy children and young adults. The results indicated that the immunogenicity of the study vaccine satisfies the Korean MFDS standards, and its safety profile was also comparable to that of the control vaccine.
Research on Korea's disease burden estimates that a total of 2,900 excess deaths associated with flu occur each year, with an all-age flu-related excess mortality rate of 5.97 per 100,000.1 Currently, there are two influenza A subtypes and two lineages of influenza B that circulate world-wide each year.8 Of these, influenza B viruses account for 20%–25% of total global influenza cases. The influenza B/Yamagata lineage has circulated every 2–4 years since the early 1940s, while the influenza B/Victoria lineage first appeared in the 1980s, when it circulated widely. In the 1990s, B/Yamagata became the more dominant lineage once again.9 Beginning in 2001, however, both B lineages have been known to co-circulate during a single season. With the exception of the influenza pandemic in 2009, influenza B viruses have accounted for 22%–44% of all cases of flu-related pediatric mortality between 2004 and 2011.101112
The most widely used seasonal influenza vaccine today is the trivalent influenza vaccine developed in the 1970s, which contains the A/H1N1 strain, A/H3N2 strain, and one influenza B lineage. Unfortunately, the previous decade saw frequent lineage mismatches between the vaccine strain and the circulating strain, resulting in reduced vaccine efficacy and consequent consumer distrust towards vaccination. The increased disease burden due to the concurrent circulation of both B lineages in a season, difficulties in predicting which of the two lineages will circulate during the upcoming flu season and manufacturing the correct vaccine to match the circulating strain, and the inadequate cross-protection provided in the case of a mismatch are good reasons for producing a quadrivalent influenza vaccine that protects against all four influenza strains, including both B lineages.13
As such, attempts to develop a safe and effective quadrivalent influenza vaccine were launched, and the first quadrivalent influenza vaccine was approved in the United States in 2012. Since the 2013–2014 flu season, the WHO and the U.S. FDA have recommended the use of quadrivalent influenza vaccines. In the U.S., there are currently 3 pharmaceutical manufacturers producing quadrivalent influenza vaccines, and the development of new vaccines is in the works.1415 In terms of approval age, split-virion vaccines have been approved for individuals 6 months and older or 3 years and older depending on the manufacturer, while live attenuated vaccines have been approved for individuals between 2 and 49 years of age. In December 2014, the MFDS approved the first Korean quadrivalent influenza vaccine for use in individuals aged 3 years and older. As of September 2016, 3 pharmaceutical manufacturers are producing quadrivalent influenza vaccines, 2 of which are split-virion vaccines and 1 of which is cell-culture-derived.1
Recommendations for quadrivalent influenza vaccines vary by country. While the WHO, Germany, the U.S., Hong Kong, Canada, Italy, France, Belgium, and Brazil tend to take a more permissive stance towards trivalent influenza vaccines relative to quadrivalent influenza vaccines, the United Kingdom and Australia actively recommend the use of quadrivalent influenza vaccines over trivalent influenza vaccines. Since 2014, Germany has recommended quadrivalent influenza vaccines over trivalent influenza vaccines for all long-distance travelers, and Brazil has also begun to recommend quadrivalent influenza vaccines for its elderly population.16 After 2012, quadrivalent influenza vaccines began to be developed, approved, and manufactured around the world, allowing for the accumulation of data pertaining to the efficacy of these vaccines. The current recommendations and guidelines regarding seasonal influenza vaccines could change based on the epidemiology of influenza viruses circulating each year.
The present clinical trial was conducted with the cooperation of 12 institutions in Korea to evaluate the immunogenicity and safety of GC3110A (split-virion quadrivalent influenza vaccine developed by Green Cross Corporation) relative to those of GC Flu pre-filled syringes (trivalent influenza vaccine) in healthy children and young adults aged ≥ 6 months to < 19 years. The ultimate aim of the study was to improve the competitiveness of Korea in the global defense effort against seasonal influenza viruses by providing a rationale for the in-house production of quadrivalent influenza vaccines that include both lineages of influenza B virus.
In this study, in the GC3110A group, the lower boundary of the two-sided 95% CI for the SCR of each subtype at day 28 following the final dose exceeded the MFDS standard of 40%. In the same group, the lower boundary of the two-sided 95% CI for the SPR of each subtype at day 28 following the final dose exceeded the MFDS standard of 70%. The quadrivalent vaccine's safety profile was comparable to that of the control (trivalent) vaccine with no notable differences. This result is consistent with those of existing quadrivalent influenza vaccine studies.1718
The age group immunogenicity subanalysis was performed with the following three age groups: ≥ 6 months to < 3 years, ≥ 3 years to < 9 years, and ≥ 9 years to < 19 years. The proportion of subjects in the GC3110A group who achieved seroconversion for each strain was confirmed to exceed 40% across all age groups. The proportion of subjects aged ≥ 6 months to < 3 years in the GC3110A group who achieved post-vaccination seroprotection based on the HI antibody titer against each vaccine strain was 82.9% for A/H1N1, 68.5% for A/H3N2, 63.1% for B/Yamagata, and 49.5% for B/Victoria, failing to meet the MFDS standard of 70%. However, results pertaining to the other age groups satisfied the MFDS standard. Based on these results, the study vaccine GC3110A meets with MFDS approval criteria for use in individuals 3 years and older.
FLULAVAL® Quadrivalent, FLUZONE® Quadrivalent, and FLUMIST® Quadrivalent are currently approved by the U.S. FDA for use in infants and children under the age of 3 years. Of these, FLULAVAL® Quadrivalent is approved in Canada, the U.S., and Mexico for use in individuals aged 6 months and older, with a single dose of 0.5 mL. FLUZONE® Quadrivalent is approved for use in individuals aged 6–35 months at a dose of 0.25 mL, while a 0.5 mL dose is approved for use in individuals aged 36 months and older. FLUMIST® Quadrivalent is an activated influenza vaccine approved for use in individuals aged 2 years and older. Citing the poor performance of influenza A(H1N1)pdm09 during the 2013–2014 and 2015–2016 influenza seasons, the Advisory Committee on Immunization Practices (ACIP) of the U.S. Centers for Disease Control and Prevention (CDC) has temporarily prohibited the use of FLUMIST® Quadrivalent in the U.S.19
There are several limitations of this study. The proportion of subjects aged ≥ 6 months to < 3 years did not meet the MFDS approval standard. Potential causes may include the small number of subjects aged ≥ 6 months to < 3 years group in relation to numbers in the other groups, as well as the dosage, which was only half of what the subjects over 3 years of age received. Taking into account the immature immune systems of young children, the dosage in this age group may have to be reconsidered. In fact, there has been a randomized control study comparing the efficacy and safety of a 0.25 mL dose and a 0.5 mL dose of a split-virion quadrivalent influenza vaccine in children aged ≥ 6 months to < 3 years. The study reported that the 0.5 mL dose was non-inferior to the 0.25 mL dose in terms of immunogenicity and safety regarding all vaccine strains and that no differences in adverse events or reactogenicities were observed. In the 6–17 months age group, in particular, the 0.5 mL dose resulted in superior defense against both lineages of influenza B compared to the 0.25 mL dose.20 Building on these results, it would be beneficial to conduct a follow-up study including a larger number of subjects aged ≥ 6 months to < 3 years, as well as a higher vaccine dosage.
Following the production of monovalent influenza vaccines in the 1930s, bivalent and trivalent influenza vaccines have been developed over the years. In 2012, the first quadrivalent influenza vaccine was approved, in keeping with epidemiological changes. Since then, many countries have adopted the use of quadrivalent influenza vaccines. The present study is the first to examine the efficacy and safety of a Korean egg-cultivated quadrivalent split-virion influenza vaccine in healthy children and adolescents.
We found that the study vaccine, GC3110A, offered immunogenicity that met the MFDS standards if used in healthy children and adolescents aged ≥ 3 years to < 19 years. Its safety profile was also found to be similar to that of the control vaccine. Based on the above results, we expect that the new quadrivalent split-virion influenza vaccine will offer broader protection to children and adolescents against seasonal influenza than the existing trivalent influenza vaccines.
 XML Download
XML Download