

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Hereditary cerebellar ataxias comprise a heterogeneous group of neurodegenerative, metabolic, and genetic disorders (1234). They are classified based on their pattern of inheritance, and about 50 genes have been identified (1234). Autosomal dominant cerebellar ataxias are typically observed in patients aged 20–50 years (123). During childhood, most of the hereditary cerebellar ataxias are autosomal recessive, X-linked, or mitochondrial (1234). Autosomal recessive cerebellar ataxias are categorized into three subclasses according to age at onset and coexistence of cerebellar atrophy: ‘Friedreich's-ataxia-like,’ ‘Friedreich's-ataxia-like with cerebellar atrophy,’ and ‘early-onset ataxia with cerebellar atrophy (12).’ However, assessment of hereditary cerebellar ataxias is confusing, as there are many chemical studies and genes to consider (1234).
The conserved oligomeric Golgi (COG) complex, which consists of 8 subunits, is an important membrane protein for maintaining the Golgi structure (56). It plays a critical role in retrograde vesicular trafficking in the Golgi apparatus (56). COG deficiency that is one of the subtypes of congenital disorders of glycosylation (CDG)-II, causes glycosylation defects and other posttranslational modifications of glycoproteins (56). In humans, mutations of the genes encoding the COG1 and COG4–COG8 subunits have been reported (56789101112131415). Since the first report in 2009, five mutations of the gene encoding COG5 have been reported in seven patients (111213). Most of these patients had hypotonia, microcephaly, and developmental delay with or without short stature (111213). Combined cerebellar atrophy was noted in two unrelated patients (111213).
The present study identified a novel heterozygous deletion of COG5 in a family with three affected siblings presenting with early-childhood-onset Friedreich's-ataxia-like phenotypes, isolated cerebellar atrophy, intellectual disability, and scoliosis. However, the patients exhibited normal growth and muscle tone differently from the previous severe cases with homozygous or compound heterozygous mutations of COG5. This finding suggests that variations of COG5 need to be considered in patients with similar early-onset Friedreich's-ataxia-like phenotypes and isolated cerebellar atrophy.
CASE DESCRIPTION
In January 2013, a girl was admitted to the hospital due to ataxic gait, scoliosis and intellectual disability. In her family, three female siblings including her (proband) showed Friedreich's-ataxia-like phenotypes with isolated cerebellar atrophy (Fig. 1A). They were born at full term to unrelated healthy parents and their perinatal history was uneventful. Their ataxia was first detected below 2 years of age in all patients. Dysmetria, dysdiadochokinesia, and dysarthria were observed with decreased deep tendon reflex. Scoliosis was detected between 1 and 4 years of age, and developmental slowing was recognized after the first year. All affected patients had mild-to-moderate intellectual disability but had no regression. The height and head growths were normal. No hypotonia, seizures, abnormal movements, ophthalmologic problems, neuronal hearing loss, facial dysmorphism, skin lesions, or abnormalities in the internal organs were observed (Table 1). Brain magnetic resonance imaging (MRI) demonstrated isolated diffuse cerebellar atrophy with enlarged interfolial spaces in a normal-sized cerebellum, although the supratentorial structures of the brain appeared to be normal in all patients (Fig. 1B). Extensive chemical, metabolic, and molecular genetic studies were performed, including a test for the gene encoding frataxin. All other tests except for whole-exome sequencing (WES) failed to establish the causes in this family.
Fig. 1
Family pedigree and brain MRI. (A) Family pedigree of the family with early-childhood-onset Friedreich's-ataxia-like phenotype and isolated cerebellar atrophy. (B) Midsagittal T1-weighted brain MRI in patients III-3 and III-4 (proband) showing cerebellar atrophy with enlarged interfolial spaces in the cerebellum. No abnormalities in other parts of brain parenchyma were noted.
MRI = magnetic resonance imaging.

Table 1
Clinical features and radiologic findings of the patients

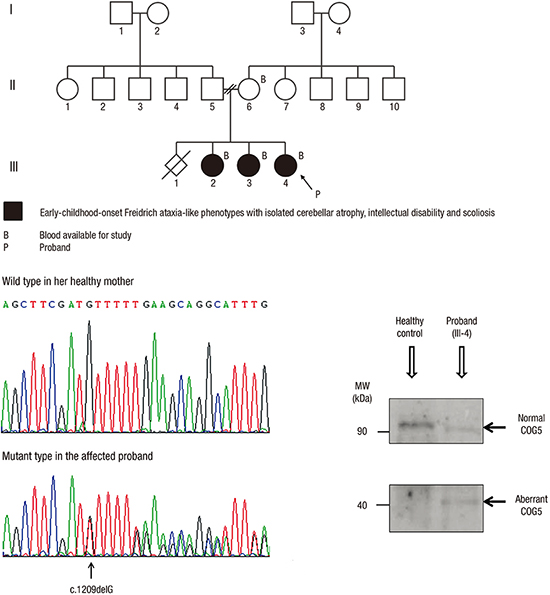
WES was performed for the three affected siblings and their mother using a TruSeq Exome Kit (Illumina Inc., San Diego, CA, USA) on a HiSeq2000 platform (Illumina Inc.). The blood sample from their father was not available. The obtained sequence reads were aligned to the human genome (hg19) using Bowtie 2. Allele frequencies of the known variants were confirmed from multiple databases, including the 1000 Genomes Project, the National Heart, Lung, and Blood Institute Exome Sequencing Project, the Single Nucleotide Polymorphism Database, and the Korean Single Nucleotide Polymorphism Database (400 Korean controls; http://nih.go.kr/NIH_NEW/main.jsp). To prioritize the variants, we established and tested our bioinformatics workflow (Fig. 2A). No potential variant was found under autosomal recessive or compound heterozygous models. Under autosomal dominant model, only one potential variant was left: the bioinformatics pipeline detected a novel heterozygous deletion in exon 12 of COG5 causing a frameshift and premature stop (c.1209delG, NM_181733.2; p.Met403IlefsX3, NP_859422.2). Sanger sequencing reverified the presence of the COG5 variation only in the patients (Fig. 2B).
Fig. 2
Mutation analysis of the gene encoding COG5. (A) Bioinformatic prioritization for variants in WES. (B) Electropherograms showing the wild-type reference sequence of COG5 in the healthy mother, and a heterozygous deletion (arrow) at the fifth base in exon 12 of COG5 causing a frameshift and premature stop (c.1209delG, NM_181733.2; p.Met403IlefsX3, NP_859422.2) in the affected proband. (C) Western blotting of COG5 skin proteins from the affected proband and a healthy control with an anti-COG5 antibody. The proband had two bands at about 90 and 40 kDa, representing the normal and aberrant COG5 proteins, respectively. The level of full-length COG5 at about 90 kDa in the proband appears to be significantly decreased compared to that in the normal healthy control showing a single meaningful band.
COG5 = conserved oligomeric Golgi complex subunit 5, WES = whole-exome sequencing, GQ = genotype quality score, GATK = Genome Analysis Toolkit, SNVs = single-nucleotide variants, MAF = minor allele frequency, ESP = Exome Sequencing Project, dbSNP = Single Nucleotide Polymorphism Database, KSNP DB = Korean Single Nucleotide Polymorphism Database, SIFT = Sorting Intolerant From Tolerant, Polyphen2 = Polymorphism Phenotyping v2, dbNSFP = Database for Nonsynonymous SNPs Functional Predictions, PhyloP = phylogenetic P value, MW = molecular weight.

To assess the expression of COG5, Western blotting of the COG5 protein was performed. The skin tissues from the affected proband and a healthy control were disrupted in liquid nitrogen with a mortar and pestle, and the proteins were extracted. Western blotting of COG5 proteins was performed using an anti-COG5 antibody (ab90301, Abcam, USA). Although a single band with a COG5 protein of about 90 kDa was detected by Western blotting of normal skin tissue as reported previously (11), two bands — one of the same size (about 90 kDa) and one smaller (about 40 kDa) — were detected in the affected proband. The intensity of the 90-kDa band in the affected proband appeared to be significantly decreased compared to that in the normal healthy control (Fig. 2C).
As there is no fundamental or curative treatment for COG5 defect, the patients were supportively treated for their scoliosis by an orthopedic surgeon.
Ethics statement
This study was approved by the Human Research Ethics Committee of Chonnam National University Hospital (IRB No. CNUH-2014-179). Informed consent to participate was obtained from the mother of the affected siblings. The biospecimens were provided by the Chonnam National University Hospital Biomedical Research Institute Biobank with informed consent, under Institutional Review Board-approved protocols.
DISCUSSION
Using the traditional classification, CDG can be subdivided into two types: CDG-I and CDG-II (56). CDG-I affects the addition of glycans to proteins and CDG-II affects processing of the protein-bound N-glycans (56). CDG-I results only in an N-glycosylation defect, while CDG-II causes a combined N- and O-glycosylation defect (56). CDG can be screened with isoelectric focusing of N- or O-glycosylated proteins such as serum transferrin or apolipoprotein CIII, respectively (56). However, isoelectric focusing is available only in some specialized clinical centers, usually takes a long time and its sensitivity and specificity are unreliable (6). Moreover, transferrin glycan analysis reveals a nonspecific pattern in most patients with the CDG-II, in contrast to those with the CDG-I (6). Due to this limitation of screening tests for CDG-II, Western blotting or genetic analysis of COG subunits in suspected patients with COG deficiency is recommended (568910). WES can be an alternative useful diagnostic method in CDG-II or COG deficiency, as it can screen for diverse variants simultaneously and rapidly with only small amounts of blood.
Deficiencies in COG subunits have been reported in patients with developmental delay, intellectual disability, growth failure, hypotonia, dysmorphic features, cerebral or cerebellar abnormalities, and feeding problems (56789101112131415). The most commonly reported defective subunit in humans is COG7, followed by COG5 (5678111213). Although the clinical features are not easy to delineate in small numbers of patients, the phenotypes appear to differ between the different defective subunits (56789101112131415). Most patients with COG7 defects died below 1 year of age, and had severe intellectual disability, growth retardation, facial dysmorphisms, hyperthermia, and congenital defects of multiple internal organs (5678). However, the patients with the other COG subunit defects showed lower mortality rates (569101112131415).
All seven previously reported patients with COG5 mutations had hypotonia, developmental delay and intellectual disability (111213). Six of them had moderate-to-severe intellectual disability; all six had microcephaly and five had short stature (111213). Only one patient with mild intellectual disability exhibited normal head and height growth (13). Two patients presented with deafness and blindness and one of them had convulsions (13). Brain MRI findings were abnormal in two patients: the one patient had global cerebral and cerebellar atrophy (13), while the other patient had diffuse cerebellar and brainstem atrophy (11). Neurogenic bladder was present in two patients (13). Contracture was noted in one patient (12). Most of the patients with COG5 mutations were from consanguineous parents and had homozygous or compound heterozygous mutations (111213).
Based on the previous diagnostic algorithm for childhood ataxia, recessive cerebellar ataxias were initially suspected in the family described herein, such as ataxia telangiectasia, infantile-onset spinocerebellar ataxia, or Friedreich's ataxia (1234). Prior to the WES study in this family, COG5 deficiency was difficult to suspect initially because the patients had no hypotonia, microcephaly, or short stature (56789101112131415). The mild phenotypes in the present family might be due to the heterozygous deletion involving only one allele, although a novel heterozygous deletion of COG5 (c.1209delG) resulted in a premature stop (p.Met403IlefsX3) causing a smaller, aberrant COG5 protein and a reduction in normal COG5 expression. Isolated cerebellar atrophy in this report is also rare radiologic finding in either of COG5 deficiency or early-childhood-onset cerebellar atrophy (41113). Associated cerebral abnormalities or abnormal signal changes in the cerebellum are reported frequently (41113). Although this type of cerebellar ataxia is not common, COG5 mutational analysis need to be considered in suitable patients showing early-childhood-onset Friedreich's-ataxia-like phenotypes and isolated cerebellar atrophy.
 XML Download
XML Download