

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Vascular calcification is a pathological accumulation of calcium phosphate crystals in the medial and intimal layers of vascular walls. Vascular calcification is closely related to metabolic diseases such as chronic kidney diseases, diabetes, and vascular diseases such as atherosclerosis (1). Pathologically, two major forms of vascular calcification have been documented, although the two types often exist in the same clinical conditions. The first type is intimal calcification, which is associated with atherosclerosis-associated lipid and cholesterol accumulation under the injured endothelium. The second type is medial calcification, also known as Mönckeberg's sclerosis, which involves deposition of minerals within the vascular smooth muscle layers (2).
Vascular calcification is widely accepted as being an active process similar to bone formation. As in bone formation, many osteoblast-like cells and intermediates are involved in vascular calcification and in other types of ectopic calcification (1). Previously, vascular calcification was thought to be a marker of atherosclerosis, but it is now believed that the pathology of vascular calcification differs from that of atherosclerosis (3). For example, vascular calcification results in adverse hemodynamic consequences such as reduced vessel elasticity and thereby stiffening of arteries, which ultimately causes an increase in pulse pressure and isolated systolic hypertension (4).
The development of calcification in patients with chronic kidney disease is closely associated with dysregulation of mineral metabolism, such as long-term elevations in the serum phosphate level as well as hypercalcemia, which leads to higher cardiovascular morbidity in these patients. Importantly, elevated extracellular calcium and phosphate levels have been shown to affect the survival and phenotype of vascular smooth muscle cells (VSMCs), leading to a pattern of cellular adaptations and damage that ultimately promotes calcification. Vascular calcification is also highly prevalent in type 2 diabetes mellitus, which contributes to worsening of cardiovascular events. Although the vascular calcification related to atherosclerosis, chronic kidney diseases, and type 2 diabetes mellitus has been well documented from a clinical aspect, the molecular mechanisms underlying the development and progression of vascular calcification remain under debate. In the present review, we give a brief overview of the current understanding of vascular calcification with respect to osteoblast-like cells and mediators in blood vessels. We then describe our new findings on the involvement of histone deacetylases (HDACs), which are epigenetic regulators, and their modifiers in the development of vascular calcification.
CURRENT VIEW OF THE MECHANISMS OF VASCULAR CALCIFICATION: OSTEOGENIC CELLS AND SIGNAL MOLECULES
Cellular components in vascular calcification
Increasing evidence suggests that vascular calcification undergoes a process similar to bone formation, which is mediated by osteoblast-like cells in situ or from the circulation. One osteoblast-like cell that has been implicated in situ is the VSMC (5). Prior to the eventual results of vascular calcification, VSMCs undergo a phenotypic switching from a contractile to a synthetic and osteogenic phenotype through the loss of contractile marker proteins such as smooth muscle α-actin and smooth muscle 22α and the gain of osteogenic proteins like osteocalcin, osteopontin, and alkaline phosphatase (6). These resulting cells can produce a mineralizing matrix that is discrete from normal vascular tissue. In addition to VSMCs in the medial layer, it is noteworthy that osteoblast-like cells can also be derived from endothelial cells in the intima, pericytes in microvessels, calcifying vascular cells, myofibroblasts in the adventitia, and progenitor cells (7).
These osteoblast-like cells can communicate with each other through different signaling mechanisms, thereby contributing to the development of vascular calcification. Thus, understanding of those pathways will provide important insights into the development of a therapeutic platform. The major stimuli for ectopic calcification in blood vessels include oxidative stress, oxidized lipid, and phosphates (8). It is noteworthy that oxidative stress also causes atherosclerosis and other cardiovascular pathologies (9) and is associated with human diseases such as diabetes mellitus and chronic kidney diseases (10). Likewise, oxidative stress can also cause an osteogenic phenotypic switching of VSMCs (11).
Apoptosis and mitochondrial dysfunction
Both viable and dead VSMCs contribute to vascular calcification; the contribution of apoptosis has been well documented in experimental models of phosphate-induced VSMC calcification (12). In addition, both viable cells and cells undergoing apoptosis are actively involved in the development of vascular calcification by releasing extracellular vesicles (13). These extracellular vesicles provide the nucleus for the deposition of calcium in the extracellular matrix. It is well known that mitochondrial dysfunction contributes to the development of atherosclerosis. Mitochondrial damage results in a reduction in ATP production and an impairment in overall cellular function. This mitochondrial damage is one of the important initial triggering signals in the early phase of atherosclerosis development (14). Considering that atherosclerosis shares many initial signal cascades with vascular calcification and that both are often found concomitantly in the same pathologic lesions, it is likely that this mitochondrial dysfunction also contributes to the development of vascular calcification. Indeed, mitochondria are involved in the intrinsic apoptosis pathway, which in turn activates caspase-9 signal via release of cytochrome c from the cytosol and thereby leads to nuclear DNA fragmentation and other apoptotic phenotypes (15).
Stress and various stimuli can induce perturbations of the endoplasmic reticulum, which results in the accumulation of misfolded and unfolded proteins and the resultant pathologic consequences of apoptosis (16). Likewise, increasing evidence has elucidated the contribution of endoplasmic reticulum stress in the development of vascular calcification in pathologic conditions with pro-calcifying stimuli, such as high glucose in type 2 diabetes mellitus (17), chronic kidney diseases (18), and atherosclerosis (19). The endoplasmic reticulum stress-induced vascular calcification is known to be mediated by several molecules, such as activating transcription factor 4 (ATF4) and C/EBP homologous protein (CHOP) (2021).
Bone morphogenic proteins (BMPs)
The BMPs, a subgroup of the TGF-β superfamily of growth factors, provide essential signals for normal developmental processes such as embryonic patterning, organogenesis, and bone formation. BMPs also work in pathological conditions including tumor angiogenesis, diabetic vascular complications, and ectopic calcification (22). Of the more than 20 BMP subtypes discovered, BMP2 is closely associated with the development of vascular calcification (23). As discovered in human atherosclerotic calcification lesions, VSMC-specific overexpression by transgenic introduction of BMP2 in apolipoprotein E (apoE)-disrupted mice accelerates vascular calcification (24). Conversely, BMP2 inhibition reduces vascular calcification in atherosclerotic low-density lipoprotein receptor (LDLR)-deficient mice (25). Calcifying phenotypic switching, a trans-differentiation of VSMCs into osteogenic cells, interestingly, is also known to be caused by BMP2 (2325), which further suggests the critical involvement of BMP2 in the development of vascular calcification.
Other signal molecules of vascular calcification
Upon binding of BMP2, both type I and type II BMP2 receptors form a heterodimeric complex, which then phosphorylates the signal proteins named small mothers against decapentaplegic (SMAD) 1/5/8 to relay the signal inside the cells. SMAD1/5/8 again makes a heteromeric complex with SMAD4 and shuttles into the nucleus to initiate transcription of the downstream genes (26). The main transcription factors in association with the BMP2/SMAD signal are runt-related transcription factor 2 (RUNX2) and muscle segment homeobox 2 (MSX2) (523). RUNX2 plays an important role in the differentiation of osteoblasts and bone formation (2728). RUNX2 is not expressed in quiescent normal vessels. Its expression is, however, highly upregulated in calcified vessels as BMP2 (2930).
Like RUNX2, MSX2 is an important transcription factor in osteoblast differentiation and bone formation (3132). In blood vessels, it was reported that BMP2 also activates MSX2 for pro-calcific effects (3334). Vascular MSX2 expression is increased in the vascular calcification in LDLR-knockout mice fed a high-fat diet (35), whereas VSMC-specific knockout of both MSX1 and MSX2 attenuates the vascular calcification in those mice (36).
Wnt/β-catenin signals are critical for mediating cell differentiation, proliferation, and death; Wnt binds to its membrane-bound receptors of Frizzled and LRP-5/6, which causes translocation of nuclear β-catenin and downstream target gene transcription (37). Wnt also targets many vascular calcification-related proteins, such as RUNX2 and osteocalcin. Like MSX-2, in response to inflammation and reactive oxygen species, the Wnt signal in the adventitia is activated by BMP2/4 released from pericytes and the endothelium (38). In addition, the Wnt/β-catenin signal induces deposition of calcium in pericytes via BMP2 (39), which suggests crosstalk between BMP and Wnt signals in the development of vascular calcification. These osteogenic proteins, BMP-2, MSX2, and Wnt, are also known to induce endoplasmic reticulum stress, which might provide an alternative pathway for their pro-calcifying effects (3440).
Anti-calcific molecules
As observed in patients with chronic kidney disease, loss of anti-calcific molecules also provides an important mechanism for the development of vascular calcification. Matrix G1a protein (MGP), fetuin-A, and osteoprotegerin (OPG) have been implicated as anti-calcific molecules (6). In addition to its direct binding to calcium to prevent vascular calcification, MGP reduces BMP2 activity by direct binding to BMP2 (41), which was further supported by the results of an in vivo animal study (42). Fetuin-A, a glycoprotein that is released from adipose tissue and liver, binds to calcium and hydroxyapatite in the blood to prevent vascular calcification (643).
The recent discovery of OPG, a phosphoprotein, and its associated proteins, receptor activator of nuclear factor-κB (RANK)/RANK ligand (RANKL), provides evidence of a direct association of vascular calcification with bone formation and loss (44). As expected, OPG knockout mice show severe osteoporosis, whereas OPG overexpression attenuates osteoclast differentiation and then causes increased bone mass (45). As expected, a decrease in OPG activity results in vascular calcification (46). Mechanistically, RANKL binds to its physiologic receptor, RANK, to induce osteoclast differentiation (47). OPG, however, interrupts the RANK/RANKL signal. This RANKL/RANK/OPG signal complex has an emerging role in the cellular communications in vascular calcification development (44). RANKL, interestingly, directly induces the release of BMP2 from human aortic endothelial cells, which suggests direct crosstalk between the two signal cascades (48).
HDACs
Protein posttranslational modifications
Proteins are not stable but are subject to active enzymatic modification, such as phosphorylation, acetylation, methylation, ubiquitinylation, carboxylation, nitrosylation, and sumoylation. These modifications, named posttranslational modifications, are finely balanced by two opposite enzymes, such as kinases vs. phosphatases, acetyltransferases vs. deacetylases, methyltransferases (methylases) vs. demethylases, etc. These paired proteins are often referred to as writers (kinases, acetyltransferases, methyltransferases, etc.) and erasers (phosphatases, deacetylases, demethylases, etc.). These modifications relay biological signals that are recognized by a series of molecules, called readers, containing particular structures such as a bromodomain extra-terminal domain. The posttranslational modifications of histone molecules are especially emphasized as epigenetic regulatory mechanisms due to their properties of inheritance of the gene expression patterns. It is noteworthy that these histone posttranslational modifications work in combinatorial ways, called the histone code, which is critical in the fine regulation of diverse cellular events (49). Interestingly, not only histones but multiple other proteins undergo posttranslational modifications, which results in the fine regulation of diverse biological and pathological functions. For example, protein acetylation takes place either at lysine residues inside the proteins or at the N-terminal of the target proteins. This lysine acetylation is associated with the regulation of enzyme activity, DNA recruitment, and transcription. Typically, histone acetylation opens the nucleosome structure, thereby enabling access of the transcriptional machinery to induce the transcription of downstream genes. In contrast, histone deacetylation represses transcription (49).
Classes of HDACs
Histone deacetylation is mediated by a family of HDACs, which consists of 18 different HDAC molecules. Based on their structural similarities and substrates, the 18 HDACs are divided into 4 classes. HDAC1/2/3/8 belong to class I, whereas HDAC4/5/6/7/9/10 are class II. The requirement for NAD+ for their deacetylase activity results in HDAC12–18 being classified as class III HDACs. The class IV HDAC group has a single member: HDAC11. In addition, class II HDACs can be divided into class IIa (HDAC4/5/7/9) and IIb (HDAC6/10). Among these HDACs, the deacetylase domain is well-conserved. Fig. 1 shows the molecular structures and tissue distributions of the HDACs.
Fig. 1
Class, molecular structure, numbers of amino acids, estimated molecular weights, and tissue distribution of the HDACs. The key to the colored box is as follows: black, HDAC domain; red, MEF2C binding domain; yellow, nuclear localization signal; blue, nuclear export signal. Molecular weights of HDACs on western blot gels are higher than expected owing to their posttranslational modifications. To simplify the diagram, the detailed structure of the class III HDAC with NAD+-dependent deacetylase activity is not shown.
HDAC = histone deacetylase, MEF2C = myocyte enhancer factor 2C.

It is somewhat in debate whether these HDACs have eventual deacetylase activity (50). Rather, as shown in the case of class IIa HDACs, some HDACs provide a scaffolding function by binding to other transcription factors including myocyte enhancer factor 2 (MEF2), thereby regulating the transcriptional activity of their binding partners (51) (Fig. 1). Some reports, including ours, however, have elucidated the existence of intrinsic deacetylase activity of the class II HDACs (5253). In the case of the class I HDACs, although the molecular sizes are relatively small and the deacetylase domain covers almost the entire structure (Fig. 1), their intrinsic activities are greater than those of the class IIa HDACs. Recently, instead of HDAC, a new term of lysine deacetylase (KDAC) is being used, because many proteins other than histones have been discovered as substrates of HDACs. This modification of nonhistone proteins provides diversity in their functions, and the acetylation status is finely regulated by protein acetyltransferases and KDACs (54).
Posttranslational modifications of HDACs
Interestingly, the HDACs themselves undergo diverse posttranslational modifications. These modifications regulate HDAC activity by altering their intrinsic enzymatic activity, their intracellular localization, their binding partners, or their protein stability. For example, regardless of their intrinsic activity, the intracellular localization of the class II HDACs provides their repressive regulatory mechanism (51). Phosphorylation causes cytosolic shuttling of the class IIa HDACs from the nucleus, which causes de-repression of prohypertrophic genes and thereby induces cardiac hypertrophy (5556). In contrast, a recent report demonstrated the opposite action of nuclear accumulation of HDAC5, a class II HDAC, in response to β-adrenergic simulation in cardiomyocytes (57). Our group also reported that either phosphorylation (58) or acetylation (52) induces activation of HDAC2, a class I HDAC, and thereby induces cardiac hypertrophy.
It is interesting that each posttranslational modification can affect other modifications and that multiple modifications in one molecule at different locations or even at a single residue (e.g., lysine) can take place. These multiple modifications may act synergistically or antagonistically. As an example of multiple modifications at a single residue, consider lysine. Lysine can be a target of acetylation, methylation, or ubiquitination. One of the well-known mechanisms of multiple modifications at a single residue is histone 3 lysine-9 (H3K9), which serves as a substrate for both acetylation and methylation. Trimethylated histone 3 lysine-9 (simply, H3K9me3) is associated with repression of transcription, whereas acetylation of H3K9 indicates an increase in transcription. The posttranslational modifications of the HDACs and the regulatory mechanisms have been reviewed previously by our group (54).
HDACS IN CARDIAC DISEASES: MECHANISTIC CONCEPTS OBTAINED FROM THE USE OF HDAC INHIBITORS
The clinical use of HDAC inhibitors as a therapeutic was first emphasized in neoplastic diseases such as cutaneous T cell lymphoma. However, their applications are now being widened to diverse diseases including cardiovascular diseases, which leads us to further investigate the fine regulatory molecular mechanisms of HDACs in homeostasis and pathology. In addition to their role as an anti-cancer drug, HDAC inhibitors are under extensive pioneering investigations for cardiac diseases such as arrhythmia (59), cardiac fibrosis (60), myocardial infarction (61), and hypertension (62).
Cardiac hypertrophy is an enlargement of individual cardiomyocytes that causes an increase in heart mass. Cardiac hypertrophy is caused by either endogenous or exogenous stresses. Congenital hypertrophic cardiomyopathy is often associated with mutations of cardiac genes (63). Besides these endogenous causes, high workload induced by exogenous stresses also results in cardiac hypertrophy. Those stresses include valvular heart diseases, uncorrected sustained hypertension, and myocardial infarction. The initial hypertrophic process is believed to be an adaptive response to those stresses. Sustained stresses caused by uncontrolled underlying diseases, however, result in pathologic hypertrophy that is accompanied by severe diastolic dysfunction and cardiac fibrosis without apparent contractile impairment. Pathologically hypertrophied hearts then enter a decompensated phase, which results in contractile dysfunction and heart failure.
Through the investigations of many groups including ours, it is known that HDAC inhibitors are beneficial in the prevention of cardiac hypertrophy and heart failure (5255565864656667686970). As for the regulation of cardiac hypertrophy, class II HDACs work as anti-hypertrophic mediators by repressing the fetal gene program as discussed above, whereas HDAC2, a class I HDAC, is prohypertrophic by decreasing the anti-hypertrophic mediators. Considering that pan-HDAC inhibitors that block both class I and class II HDACs can efficiently block cardiac hypertrophy, class I HDAC may contribute to the overall HDAC activity in the heart. As described above, the class II HDAC activity is much lower than that of the class I HDACs. It has been reported that the anti-hypertrophic action of the class II HDACs is independent of their deacetylase activity (64), whereas we reported that minute but significant activity of HDAC5, a class II HDAC, is important to its anti-hypertrophic properties mediated by deacetylase of HDAC2 (52). In addition to the fundamental roles of HDAC2 in the development of cardiac hypertrophy, we elucidated that prohypertrophic HDAC2 activation is mediated by its posttranslational modification (5258) or by its binding partner heat shock protein 70 (69).
ROLE OF HDACS IN VASCULAR DISEASES
The role of HDACs in vascular diseases was also basically investigated by utilization of HDAC inhibitors. For example, it is known that HDAC inhibitors have an anti-angiogenic effect that lends them additional anti-cancer properties (71). Based on this usage of HDAC inhibitors, it was discovered that class II HDACs work as pro-angiogenic factors. Interestingly, however, a positive correlation between HDAC inhibitors and angiogenesis has also been reported (7273).
Atherosclerosis, a narrowing of the blood vessels, is characterized by chronic inflammation, accumulation of lipids, a fibrous cap, and VSMC proliferation. Atherosclerosis in the coronary artery especially is the most common cause of myocardial infarction. Many research groups including ours have previously reported that non-class-specific HDAC inhibitors prevent neointimal proliferation (7475). By contrast, as in the case of angiogenesis, it has also been reported that HDAC inhibitors exaggerate atherosclerosis (76). This discrepancy might be caused by the duration of drug treatment or the initiation point of administration of the inhibitors. In addition, the diversity of HDAC structures and subtypes contribute to the opposite effects of HDAC inhibitors in atherosclerosis or angiogenesis.
HDAC1 AND ITS POSTTRANSLATIONAL MODIFICATION IN VASCULAR CALCIFICATION
In general, including their anti-neoplasmic properties, it is widely accepted that HDAC inhibitors have beneficial effects as therapeutics for epilepsy, angiogenesis, cardiac hypertrophy/heart failure and fibrosis, and myocardial infarction as described above. We first postulated that HDAC inhibitors may also have a beneficial function to alleviate vascular calcification. Thus, using methods to evaluate vascular calcification, we performed a preliminary study utilizing HDAC inhibitors. On the contrary, however, we found that HDAC inhibitors potentiated vascular calcification. This finding was quite interesting because it suggested new possible adverse effects in the application of HDAC inhibitors in other diseases as well as possible novel pathways regarding HDAC in vascular calcification. Indeed, during our work, one research group published that HDAC inhibitors worsen vascular calcification by activating RUNX2 (77).
Role of HDAC1 in vascular calcification
We first observed that treatment with either trichostatin A (a non-class-specific HDAC inhibitor) or apicidin (a class I-specific HDAC inhibitor) did not affect calcium content in primarily cultured VSMCs (78). Interestingly, however, trichostatin A and apicidin potentiated vascular calcification induced by either inorganic phosphate in VSMCs or vitamin D3 in mice. In the following series of experiments, we found that loss of activity of HDAC1, one of class I HDACs, results in the exaggeration of vascular calcification. Specifically, we found that 1) HDAC1 siRNA imitated the effects of HDAC inhibitors, and 2) VSMC-specific deletion of HDAC1 in the mice exhibited enhancement of vascular calcification phenotypes. We further examined the expression levels of class I HDACs (HDAC1, HDAC2, HDAC3, and HDAC8) and found that vascular calcification reduces the protein expression level of HDAC1. Indeed, in vascular calcification samples obtained from diverse animal models or from human calcified coronary artery samples, we found that the expression of HDAC1 was significantly reduced (78).
Proteasomal degradation of HDAC1 in vascular calcification
We next questioned how vascular calcification causes the reduction in HDAC1 expression. Through a series of experiments, we found that HDAC1 protein degradation was enhanced in vascular calcification conditions. These results raised the possibility of involvement of the protein-degradation pathway in the reduction of the HDAC1 expression level. Indeed, we found that the polyubiquitination-dependent protein degradation pathway is involved in the degradation of HDAC1 in vascular calcification events. Together with the autophagic degradation system, proteasomal degradation is the most important system of the protein quality control pathways to remove abnormal or unnecessary proteins and recycle them. In the proteasomal degradation pathway, polyubiquitination of proteins is the key initial signal to recognize the target to be degraded (79). As exemplified in the catabolic process during muscle atrophy, these ubiquitin-proteasome and autophage-lysosome pathways are very important in both pathophysiology and normal homeostasis (80).
Polyubiquitination is caused by the covalent bonding of target proteins with a serial conjugation of small proteins named ubiquitin (81). Of note, polyubiquitination by multiple conjugations of ubiquitin K48 initiates 26S proteasome-dependent degradation, while K65 polyubiquitination as well as monoubiquitination (attachment of a single ubiquitin molecule to a target) is not associated with protein degradation but is involved in endocytosis, intracellular shuttling, enzymatic activation, or transcriptional regulation (82).
To induce polyubiquitination-dependent protein degradation, the target protein should undergo sequential conjugations catalyzed by serial enzymes of E1, E2, and E3 ligases (83). E1 and E2 ligases are involved in a common pathway; they work to make the multiple conjugations of ubiquitin proteins themselves. Then the E3 ligases determine the target protein and make the final conjugation of the multiple ubiquitin molecules generated by the E1/E2 ligases to the target (83). Several hundreds to thousands of E3 ligases are expected to be involved in the final process. Because target protein specificity is determined by the E3 ligase, it is important to find which E3 ligase is involved in the proteasomal degradation pathway of a protein of interest. Likewise, we believed that finding an HDAC1-specific E3 ligase was critical for further investigation of the HDAC1-associated mechanism in the development of vascular calcification.
MDM2, an HDAC1 E3 ligase, exaggerates vascular calcification
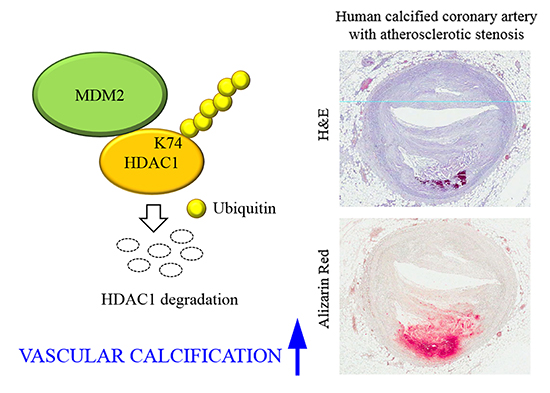
Thus, we next tried to find the HDAC1 E3 ligase involved in vascular calcification. First, by literature searching, we found four HDAC1-specific E3 ligases: CHRF (84), MDM2 (85), PIRH2 (86), and Cullin3-REN (87). In addition, we performed a cDNA microarray and searched for E3 ligases that were upregulated by treatment with inorganic phosphate. In both the literature search and the cDNA microarray, the expression of MDM2 was dramatically increased (78). MDM2 has multiple targets to induce polyubiquitination and was first elucidated as a p53-specific E3 ligase (88). Because of its property to antagonize p53 activity (88) and the observation of an increase in its expression in cancer (89), MDM2 is considered an oncogene that serves as a clinical biomarker in certain cancers. MDM2 has multiple targets, of which HDAC1 is one (85).
Next, we further confirmed that MDM2 is involved in an HDAC1-associated vascular calcification mechanism as an E3 ligase by obtaining the results of 1) MDM2-upregulation by inorganic phosphate, 2) MDM2-induced increase in calcium contents in VSMCs, 3) direct binding of MDM2 to HDAC1, 4) MDM2-induced degradation of HDAC1, and 5) MDM2 RING domain-dependent ubiquitination of HDAC1. In addition, we observed that MDM2 is upregulated in diverse animal models and in human coronary samples with either medial calcification or intimal calcification (78). The most striking observation in our work is the attenuation of vascular calcification by RG 7112, an MDM2 inhibitor (78). The working hypothesis of vascular calcification regarding MDM2/HDAC1 is shown in Fig. 2.
Fig. 2
Diagram of the working mechanism of HDAC1 and MDM2, an HDAC1-E3 ligase, in the development of vascular calcification. In the healthy condition, HDAC1 inhibits osteogenic gene expression including that of RUNX2 in VSMCs. With calcification stresses, MDM2 expression is increased in VSMCs, resulting in the polyubiquitination-dependent proteasomal degradation of HDAC1. The reduced HDAC1 protein amount then causes the de-repression of calcifying genes, followed by an increase in calcium deposition in VSMCs. Therapeutic use of HDAC inhibitors in various diseases may inhibit the deacetylase activity of HDAC1, thus causing unwanted vascular calcification. In contrast, either treatment with RG 7112, an MDM2 inhibitor, or interruption of the polyubiquitination of HDAC1 may be beneficial in preventing vascular calcification (78).
HDAC = histone deacetylase, RUNX2 = runt-related transcription factor 2, VSMC = vascular smooth muscle cell.
Our findings are of major significance for several reasons. First, we have demonstrated that polyubiquitination-dependent protein degradation induces vascular calcification and that MDM2/HDAC1 signal cascades are involved in the cardiovascular pathophysiology. To our knowledge, only two E3 ligases demonstrated by two research groups including ours are known to be involved in the polyubiquitination-associated vascular calcification: MDM2 (78) and NEDD4 (90). Second, we have shown that HDAC inhibition potentiates vascular calcification. Considering that many HDAC inhibitors are being developed and are under extensive investigation as anti-cancer drugs, the likeliness of an increase in vascular calcification by HDAC inhibitors should be noted as a potential adverse effect. Third and most importantly, we have provided new concepts for drug development against vascular calcification by demonstration of the beneficial effect of blockade of either HDAC1 degradation or MDM2 activity. Additionally, RG 7112 and its associated drugs would be excellent models.
AN ALTERNATIVE EPIGENETIC REGULATION OF VASCULAR CALCIFICATION: NONCODING RNA
Through years of biochemical and bioinformatics research, diverse types of noncoding RNAs that do not encode proteins have been discovered. These novel classes of RNAs include microRNAs, long noncoding RNAs, and circular RNAs (91). Among these noncoding RNAs, microRNAs suppress the expression of target genes by sequence-specific binding to the 3'-untranslated region of target mRNAs (92). Owing to their involvement in the pathogenesis of various diseases and tissue-specific expression pattern, extensive studies are underway to use microRNAs as therapeutic targets and disease markers (93). Until now, only about a dozen microRNAs had been shown to be involved in vascular calcification (9495). Our group found that miR-124 inhibits the proliferation of VSMCs by targeting S100 calcium-binding protein A4 (S100A4), which has an important role in the proliferation of VSMCs and correlates with mineralization (9697). In addition, our computational prediction suggests that MDM2 could also be a potential target of miR-18a. As more research is ongoing to analyze the function of microRNAs in diverse systems, it is possible that additional microRNAs will be included in the list of vascular calcification-related microRNAs.
Compared to microRNAs, knowledge of other types of noncoding RNAs in vascular calcification is limited. Only one study reported the involvement of long noncoding RNA in vascular calcification. One of the famous noncoding RNAs, HOTAIR, which is extensively studied in the field of developmental epigenetics, is also implicated in aortic valve calcification (9899). It was found that suppression of HOTAIR increases the expression of calcification genes. However, no other study has reported the involvement of long noncoding RNAs or other types of noncoding RNAs in vascular calcification. Since the roles of noncoding RNAs in various experimental models are being studied extensively, it is likely that the involvement of diverse types of noncoding RNAs in vascular calcification will be revealed in the near future.
CONCLUSION
Vascular calcification is now believed to be an active process, not a passive response to the death of VSMCs. In the current review, we briefly summarized recent concepts concerning the main mechanism of vascular calcification and added our new findings of the involvement of MDM2/HDAC1-mediated polyubiquitination/epigenetic mechanisms in the development of vascular calcification. Because vascular calcification is one of the forms of ectopic calcification in the body, its intracellular mechanisms are quite similar to those regulating osteoblast. Indeed, the molecules described here, such as BMP, SMAD, RUNX2, MSX2, Wnt/β-catenin, OPG, RANK, and RANKL, are famous in the calcification of bone and tightly affect the biological behaviors of osteoblasts and osteoclasts. Thus, to our sense, it seems that much research into the mechanisms of vascular calcification is following previously established theories in bone. Then, the fundamental question becomes, what causes the osteogenic differentiation of VSMCs? That is, how is it that VSMCs have bone-forming properties? Direct stimulation by the above molecules would also cause the trans-differentiation of VSMCs into osteoblast-like cells. Or, unknown pathways that are specific to VSMCs may actively participate in the process. Thus, understanding of those signals and the associated disease states that trigger the process will provide an excellent platform for the future development of drugs to treat vascular calcification.
 XML Download
XML Download