

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Cystic fibrosis (CF), an autosomal recessive inherited multisystem disorder, is the most common life-limiting genetic disorder in Caucasians (12). Mutations of the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR) protein result in malfunction of a protein kinase A-activated chloride- and bicarbonate-selective ion channel involved in salt and water transport in multiple organs, including the lungs (3). Over 2,000 mutations have been identified (4). CF is diagnosed based on a clinical presentation of the disease and CFTR gene dysfunction. The sweat chloride test should be conducted first, followed by genetic analysis of CFTR and physiologic tests for CFTR defects (4).
Respiratory failure is the most frequent cause of death and impairment due to CF (5). Most patients with CF eventually develop irreversible end-stage bronchiectasis and obstructive lung disease, at which point conventional medical treatments are ineffective, and lung transplantation is the only therapeutic option (6). CF is the most frequent indication for lung transplantation in children (2).
According to one review, transplantation is often associated with serious complications and might not prolong life or significantly improve the quality of life. In that review, pre-transplantation colonization with Burkholderia cepacia, diabetes, and older age reportedly impaired post-transplantation survival (7). However, a later analysis suggested that lung transplantation was beneficial in patients with CF in the modern era with the implementation of the lung allocation score (5). Since the introduction of the lung allocation score, 1- and 5-year survival rates of pediatric patients have improved who underwent lung transplantation for CF (8).
CF is rare in Asian populations; an epidemiological study found that the incidence of CF was about 1 in 350,000 in Japan (9). CF is rarer in Korea, where there have been few cases of genetically-confirmed CF (10). Here, we report the case of a 12-year-old Korean girl with CF who underwent successful lung transplantation. To the best of our knowledge, she is the first Korean child with CF and a CFTR mutation to have received this treatment.
CASE DESCRIPTION
A 7-year-old girl with CF was admitted to our hospital with fever and greenish sputum. She had been diagnosed with CF 3 years previously at another tertiary hospital after treatment for recurrent Pseudomonas aeruginosa pneumonia and malnutrition. At that time, average sweat chloride concentrations on both forearms were 78.3 mmol/L on the first test and 99.0 mmol/L on the second test (reference interval: < 40 mmol/L, borderline: 40–60 mmol/L). CFTR gene mutation was positive, with c.1322T>C (p.Leu441Pro). Abdominal computed tomography (CT) scanning showed fatty infiltration of the liver and severe pancreatic atrophy. After the diagnosis, she was admitted yearly with pneumonia, and from the age of 6, P. aeruginosa and Staphylococcus aureus were isolated from her sputum. She was a full-term baby with a birth weight of 2.4 kg, born by vaginal delivery without any perinatal problems. At 3 months of age, she was treated for cytomegaloviral pneumonia and hepatitis. She has a healthy brother 5 years older than her, and the remaining family history is also unremarkable.
On admission, her vitals were: blood pressure 110/60 mmHg, heart rate 108 beats/min, respiratory rate 28/min, SpO2 100% on room air, and body temperature 38.6°C. Her height was 116.5 cm (25th percentile) and body weight was 17 kg (5th percentile) after being on growth hormone therapy for 1 year because of growth failure (her height was initially the 3rd–5th percentile with body weight below the 3rd percentile). She did not look particularly ill and was alert. Her chest expanded symmetrically without intercostal retraction. Coarse breath sounds with crackles were noted bilaterally. Her liver and spleen were not palpable. There was no clubbing or aquagenic palmoplantar keratoderma. Her total leukocyte count was 13,300/μL. C-reactive protein level was elevated at 25.01 mg/dL. Her sodium and chloride levels were in the normal range, and her HbA1c was 6.6%. Chest X-ray showed bronchiectasis and multifocal nodular opacity with peribronchial infiltration in both lungs (Fig. 1A). Chest CT showed bronchiectasis in both lungs and consolidation in the right upper lobe and anterior basal segment of the left lower lobe (Fig. 1B). Abdominal CT scanning revealed severe fatty infiltration in the liver and severe pancreatic atrophy with no bile duct dilatation or nephrolithiasis. Her forced expiratory volume in 1 second (FEV1) was 1.14 L (91% predicted). Methicillin-resistant S. aureus (MRSA) and ceftazidime-resistant P. aeruginosa were isolated from her sputum. The CFTR gene mutation test was repeated, and a heterozygous T to C transition was identified (c.1322T>C), but mutation of the opposite allele was not detected (Fig. 2). Her healthy brother also had the heterozygous point mutation. No targeted genetic testing was done on her parents in our hospital.
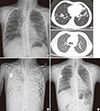 | Fig. 1Serial follow-up of radiologic findings before and after lung transplantation. (A) Initial chest X-ray at first admission to our hospital showing bronchiectasis and multifocal nodular opacity with peribronchial infiltration in both lungs (at the age of 7 years). (B) Chest CT scan taken at first admission showing bronchiectasis and consolidation in the right upper lobe and anterior basal segment of the left lower lobe (at the age of 7 years). (C) Chest X-ray taken one day before lung transplantation showing an increased extent of consolidation in both lungs. At that time, saturation was not maintained at 100% oxygen via a ventilator and ECMO (SpO2 75%–80%, pO2 35–40). (D) Follow-up chest CT scan taken 5 months after lung transplantation. (E) Follow-up chest X-ray at 2 years and 6 months after lung transplantation showing no remarkable findings.
CT = computed tomography, ECMO = extracorporeal membrane oxygenation.
|
 | Fig. 2Genotyping of CFTR. A heterozygous T to C transition was identified (c.1322T>C), but mutation of the opposite allele was not detectable.
CFTR = cystic fibrosis transmembrane conductance regulator.
|
After her first admission to our hospital, she visited our clinic regularly and was hospitalized several more times for recurrent pneumonia. From age of 9 years, carbapenem-resistant P. aeruginosa (CRPA) and methicillin-susceptible S. aureus (MSSA) were isolated along with B. cepacia. She was treated with albuterol, hypertonic saline 7% inhalation, tobramycin nebulizer every other 4 weeks, oral azithromycin 3 times a week, percussion vest, and vitamin D supplements. FEV1 declined to 0.35 L (18% predicted) by the age of 12, and pneumonia became more frequent.
At age of 12 years, she was hospitalized with a prolonged high fever, aggravation of a productive cough, and greenish sputum. B. cepacia complex (Burkholderia arboris) and CRPA were isolated from her sputum. Progression to end-stage lung disease resulted in intubation on hospital day 21 and registration with the Korean Network for Organ Sharing. Despite high-frequency oscillatory ventilator care with 100% fraction of inspired oxygen, desaturation and hypercapnia persisted. Therefore, venovenous extracorporeal membrane oxygenation (ECMO) via the left femoral vein and right internal jugular vein was applied on hospital day 25. However, respiratory failure progressed. Saturation was not maintained with 100% oxygen via the ventilator and ECMO (SpO2 75%–80%, pO2 35–40), and her chest X-ray showed increased consolidation in both lungs (Fig. 1C). On the 7th day of ECMO, she received a double lung transplant from a 10-year-old girl with brain death due to encephalitis of unknown origin. The donor-to-recipient body weight ratio was 1.5 (31.9/21 kg). At surgery, diffuse moderate-to-severe pleural adhesions were evident on the upper medial side of the recipient's left pleural cavity. Gross findings for both lungs were total consolidation and fibrosis (Fig. 3). The main bronchi of the recipient were 25% larger than those of the donor. The anastomoses sites of the main bronchi were patent post-transplantation. She was disconnected from the venovenous ECMO in the operating room. Histological examination of the explanted lungs revealed damage with multifocal abscesses and multifocal organizing pneumonia.
 | Fig. 3Gross findings of the explanted lung of the recipient showing dilated bronchioles and diffuse fibrotic lung parenchyma with atrophy.
|
She was extubated on postoperative day 8, but 6 days later she was reintubated because of hypercapnia caused by a mucous plug in a bronchus and impaired coughing. She was extubated a second time on post-transplantation day 28. However, the bilateral multiloculated pleural effusion gradually increased. Although chest tubes were inserted bilaterally and urokinase was instilled, she had to be intubated again on post-transplantation day 48. Chest tubes were repeatedly inserted and removed for multiloculated pleural effusion. Tracheostomy was performed on post-transplantation day 52, and on post-transplantation day 63, she was transferred to a general ward with a home ventilator. The chest tubes were removed after the pleural effusion resolved. On post-transplantation day 164, the tracheostomy tube was removed, and the tracheostomy stoma was closed.
Despite lung transplantation, B. cepacia and CRPA continued to be isolated from sputum culture from day 1 post-transplantation. B. cepacia was also isolated from pleural fluid and the central venous line. Intravenous antibiotics were maintained, and the central venous line was removed. At 4 months post-transplantation, B. cepacia had been eradicated from her sputum.
Because of abdominal pain and vomiting from about post-transplantation day 50, she was fed via a transpyloric tube. She also had to receive a Botox injection and balloon dilatation for pyloric spasm at 5 months post-transplantation. Her body weight was 18.7 kg at admission, 19.2 kg on the day of transplantation, 17.9 kg at 1 month post-transplantation, 16.9 kg at 2 months post-transplantation, and 24 kg at discharge. Since lung transplantation, she has required insulin therapy, perhaps because of aggravation of the pancreatic insufficiency of CF. At 3 months post-transplantation, her glomerular filtration rate declined, and the dosages of medications including immunosuppressants and antiviral agents were adjusted.
At 8 months post-transplantation, her Epstein-Barr virus (EBV) polymerase chain reaction (PCR) titer increased to 4.82 log copies/mL and remained at about 4 log copies/mL for 3 months. At that time, positron emission tomography (PET) revealed no evidence of post-transplant lymphoproliferative disease (PTLD). She received monoclonal anti-CD20 antibody (rituximab, Rituxan®; Genentech, Inc., South San Francisco, CA, USA), and the EBV PCR titer underwent negative conversion. However, from 15 months post-transplantation the EBV PCR titer again increased gradually and has waxed and waned around 4 log copies/mL to the present day without any clinical manifestations.
After lung transplantation, her pulmonary function test (PFT) gradually improved. The latest PFT (3 years post-transplantation) showed an FEV1 of 1.89 L (77% predicted) (Fig. 4). A follow-up chest CT scan at 5 months after lung transplantation (Fig. 1D) and follow-up chest X-ray at 2.5 years yielded no remarkable findings (Fig. 1E).

DISCUSSION
There are few cases of CF in Korea. The spectrum of CFTR gene mutations in Koreans seems to differ from that in Caucasians. In Caucasians, the CFTR mutation p.F508del accounts for approximately 70% of mutations. A study of the spectrum of CFTR mutations in Korean patients with CF showed no cases of the p.F508del mutation and only one example of the 32 common mutations on the screening panel constructed for Caucasians (10). Different mutations of CFTR are associated with varying degrees of severity. Our patient had a heterozygous mutation of c.1322T>C (p.Leu441Pro), which has not been previously reported. Some children with typical CF manifestations are found to have 1 CF mutation and no detectable mutation on the sister chromosome (2). Her brother was heterozygous for the same CFTR mutation but was a healthy carrier.
Respiratory failure is the major cause of morbidity and mortality in CF. Despite improved treatments and increased survival, lung transplantation is an important treatment for end-stage lung disease (11). Transplantation indications are percent predicted FEV1 value below 30%, frequent hospitalization, and refractory hypoxemia or hypercapnia (12).
CF is the most common indication for pediatric lung transplantation worldwide. Reportedly, 892 lung transplants were performed in children with CF from January 2000 to June 2015. Half the 6- to10-year-olds and two-thirds of the 11- to 17-year-olds had CF (13). However, there were regional variations. In Europe, 66% of pediatric lung transplant recipients had CF compared to 49% in North America and only 40% in the remaining geographic regions (13). In Japan, only 0.6% of lung transplant recipients had CF (14).
Lung transplantation for CF was first performed as a combined heart-lung transplant in 1983 (15). The first lung transplant in a Korean child was also a combined heart-lung transplant, in 2011. The patient was diagnosed with pediatric interstitial lung disease caused by a humidifier disinfectant (16). However, lung transplantation for CF had not been performed in Korea prior to this report.
The median survival after lung transplantation reportedly was 5.4 years in children and 5.8 years in adults. Major causes of death in the first month after transplantation were graft failure, cardiovascular problems, infection, and multi-organ failure. After the 1 year post-transplantation, obliterative bronchiolitis/bronchiolitis obliterans syndrome was the leading cause of death (13).
B. cepacia and CRPA were isolated from our patient's sputum cultures before and after transplantation. B. cepacia is associated with more rapid respiratory disease progression and a more rapid FEV1 fall. Patients with B. cepacia complex infection also have less favorable post-transplantation outcomes (7). The mortality rate was reported to be higher in the B. cenocepacia group than in other serotypes (17). In our case, B. arboris was identified by serotyping the B. cepacia complex, and at the time of transplantation, body mass index (BMI) was 12.0 kg/m2. According to one study, a low BMI may be related to chronic colonization by the B. cepacia complex (9). By 4 months post-transplantation, B. cepacia was eradicated from our patient's sputum.
Nutritional status is an important predictive factor for survival in CF (18). For patients with CF, an intake of 120%–150% of the estimated energy requirement is recommended. Our patient had feeding difficulties and required continuous tube feeding for 6 months post-transplantation. For 2 years before transplantation, she failed to gain weight. At 6 months post-transplantation, her growth started to catch up, but her height and body weight were below the 3rd percentile on the final outpatient clinic follow-up day.
One year post-transplantation, the most common complications are hypertension (> 40%), diabetes mellitus (21%), and bronchiolitis obliterans syndrome (11.2%). Five years post-transplantation, 30% have chronic kidney dysfunction (13). Our patient took amlodipine for hypertension until 8 months post-transplantation, and she still requires insulin therapy because of complications and pancreatic atrophy. She also has stage 3 chronic kidney disease.
We reported the first successful lung transplantation in a Korean child with CF. Although she has diabetes mellitus and chronic kidney disease, she has a better quality of life and prolonged life expectancy. Lung transplantation is an option for appropriately selected pediatric patients with end-stage lung disease due to CF.
 XML Download
XML Download