

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Hypocomplementemic urticarial vasculitis syndrome (HUVS) was first described in 1973 by McDuffie et al. (1). As indicated by its name, it manifests with urticaria and/or angioedema as well as a variety of systemic involvements with a low complement level. Unlike simple urticaria that is mostly due to a type I hypersensitivity reaction, HUVS is recognized as an autoimmune disease involving a type III hypersensitivity reaction (2). In 2014, a unique Korean case of HUVS with membranoproliferative glomerulonephritis was reported whose clinical course was combined with arthropathy and cardiac valvulopathy (3). Herein, we report another Korean case of HUVS with membranous nephropathy and myositis that was successfully treated with a combination of glucocorticoid and immunosuppressive agents.
CASE DESCRIPTION
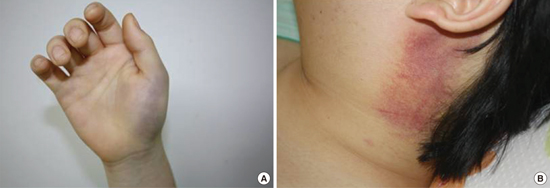
A 15-year-old child presented to our hospital in March 2010 complaining of a 2nd episode of facial swelling. About 2 weeks previously, numerous pruritic wheals and subsequent bruises developed on both hands and the abdomen (Fig. 1A). Subsequently, he noticed his lips and tongue had swollen and felt muscle weakness and pain, especially in both shoulders and thighs, that started a day before this presentation. The patient had been otherwise well with a history of tonsillectomy until the previous year when an episode of urticaria occurred 2 weeks after a swine-origin influenza A virus infection. Approximately 3 months before this presentation, he had experienced swelling of both hands and his face.
Fig. 1
Skin changes in the patient. (A) Bruise on the left hand. (B) Subsequent erythematous change with diffuse swelling on the left neck.

He was admitted under suspicion of a systemic disease. On examination, the blood pressure was 140/70 mmHg, the pulse 140 beats per minute, the temperature 37.5°C, and the respiratory rate was 22 breaths per minute. There was a diffuse swelling of both cheeks, the lips, tongue, throat, and neck. Blood levels of urea, creatinine, electrolytes, and coagulation tests were normal while those of white blood cells (WBC), erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), and muscle enzymes were elevated as follows; WBC 13,140/uL, ESR 23 mm/hr, CRP 8.4 mg/dL, creatine kinase (CK) 5,160 U/L (60–220), myoglobin 1,061.7 ng/mL (17.4–105.7). The urinalysis revealed 0 to 1 red cells and 0 to 1 white cells per high-power field and no albumin on a dipstick test. The auto-antibody (antinuclear antibody [ANA], anti-double-stranded DNA, anti-Sm, cytoplasmic anti-neutrophil cytoplasmic antibodies, and perinuclear anti-neutrophil cytoplasmic antibodies) panels were negative. C3 (87.6 mg/dL) was slightly lower than the lower limit of the normal range (88–201 mg/dL), while C4, C1q, and CH50 levels were not below the normal range. Electromyography (EMG) showed low-amplitude short-duration motor unit action potentials and magnetic resonance imaging (MRI) showed diffuse myopathy in both thighs. On the 2nd day, his body temperature rose to 38.5°C and purpura developed on his chin and neck causing tenderness (Fig. 1B). He also reported an itching sensation over the urticarial rashes and pain in both extremities. On the 4th day, subconjunctival hemorrhage developed spontaneously in both eyes without any history of trauma.
The recurrent angioedema and urticaria led us to perform a skin biopsy in the right forearm. The pathologic examination revealed a perivascular infiltration of neutrophils and lymphocytes that was consistent with leukocytoclastic vasculitis (Fig. 2A and 2B). Thus, the diagnosis of urticarial vasculitis was made. Furthermore, it was reasonable that inflammatory myositis was considered on the basis of proximal muscle weakness, elevated muscle enzymes, and myopathic pattern on EMG, although a muscle biopsy was not performed. Because the angioedema was worsening to the point that the patient felt difficulty swallowing and breathing, intravenous methylprednisolone pulse therapy (1,000 mg/day; Predisol®; Reyon Pharm Co., Ltd., Seoul, Korea) was administered for 5 consecutive days. It was followed by oral methylprednisolone (Methylon®; Alvogen Korea, Seoul, Korea) which was tapered with the addition of azathioprine (Azathioprine PCH®; Handok Teva, Seoul, Korea) and hydroxychloroquine (Oxiklorin®; Kyung Poong Pharma Co., Ltd., Seoul, Korea) as his symptoms subsided.
Fig. 2
Skin punch biopsy from a petechial area of right arm. (A) Moderate inflammatory cell infiltrates composed of neutrophils and lymphocytes involving the superficial dermis (H & E stained; original magnification × 100). (B) High-power view highlights leukocytoclastic vasculitis in the dermal vasculature (H & E stained; original magnification × 200).
H & E = hematoxylin and eosin.

After discharge, he was maintained on fexofenadine (Allegra; Handok Pharmaceutical Co., Ltd., Seoul, Korea), azathioprine (50 to 100 mg/day; Handok Teva), hydroxychloroquine (200 to 400 mg/day; Kyung Poong Pharma Co.,Ltd.), and methylprednisolone (8 to 64 mg/day; Alvogen Korea). Unfortunately, he experienced several bouts of repeated facial swelling, urticaria, myalgia, and muscles weakness with various degrees of hypocomplementemia. During each episode, he received either intravenous methylprednisolone (Reyon Pharm Co., Ltd.) pulse therapy or a reescalation of its oral dose.
Sixteen months after his initial admission, proteinuria (922 mg/day) developed without any evidence of hematuria. Thus, a renal biopsy was performed in September 2011. The biopsy specimen showed 9 glomeruli on light microscopy, none of which demonstrated any glomerulosclerosis or inflammatory cell infiltration (Fig. 3A). Both mesangial cell proliferation and glomerular capillary wall thickening were focal and segmental. An immunofluorescence assay showed mild staining for immunoglobulin (Ig) G and C3 (Fig. 3B and 3C) and no staining for IgA, IgM, and C1q along the glomerular capillary wall. On electron microscopy, the glomerular basement membrane was thickened with overlying podocyte foot process effacement. In addition, subepithelial electron-dense deposits were noted (Fig. 3D). These findings were diagnostic of membranous nephropathy. Thus, telmisartan (Pritor®; GlaxoSmithKline plc., Brentford, United Kingdom) was added for proteinuria and azathioprine (Handok Teva) was switched telmisartan (Pritor®) with maintenance of hydroxychloroquine (Kyung Poong Pharma Co.,Ltd.) and prednisolone (Solondo®; Yuhan Corporation, Seoul, Korea). On follow-up, proteinuria demonstrated complete remission but occasionally reappeared and resolved again without any increase of serum creatinine. C3 and C4 levels fluctuated with a lowest level of 70.8 mg/L and 15.4 mg/dL, respectively, whereas C1q has remained within the normal range. Thereafter, immunosuppressive agents were switched several times because of the relapse and aggravation of the cutaneous and musculoskeletal symptoms. At present, dapsone (50 mg/day; Taiguk Pharm Co.,Ltd., Seoul, Korea), tacrolimus (2 mg/day; Chong Kun Dang Pharmaceutical Corp., Seoul, Korea), and methylprednisolone (8 mg/day; Alvogen Korea) are prescribed.
Fig. 3
Renal biopsy findings. (A) Mild glomerular capillary wall thickening with normal cellularity and patent lumen (PAS stain; original magnification × 400). (B) Immunofluorescence staining for IgG along the glomerular capillary wall (original magnification × 200). (C) Immunofluorescence staining for C3 along the glomerular capillary wall (original magnification × 200). (D) A number of subepithelial deposits are observed with foot process effacement and basement membrane thickening (electron microscopy; one length of black or gray scale bar represents 1 µm).
PAS = periodic acid-Schiff, IgG = immunoglobulin G.

DISCUSSION
The patient we described had a low complement level and systemic involvement presenting with glomerulonephritis, subconjunctival hemorrhage, and myositis, although the myositis was not biopsy-proven. This met the diagnostic criteria for HUVS proposed by Schwartz et al. (4) in 1982.
It has been postulated that the immune complex plays a critical role in the pathogenesis of HUVS leading to a low complement level (2). The immune complex is the first trigger that activates the classical complement pathway by binding to C1q, leading to complement consumption and formation of the membrane attack complex. Immunostaining reveals deposition of Ig and complement along blood vessels in most patients, which support this hypothesis (2). The intermittent low C3 and C4 levels in our patient reflect the activation of the classical pathway.
The antigens eliciting the formation of antibodies are presumed to be viruses and, in some cases, medications. In this case, a swine-origin influenza infection could be the trigger considering that the influenza infection preceded the initial episode of urticaria by 2 weeks. In addition, the collagen-like region of C1q can induce antibody formation, possibly by exposing new epitopes, during the process of apoptosis (5). C1q autoantibody was found in 55% of patients in a recent French nationwide study (6). It can boost the activation of the classical complement pathway in systemic lupus erythematosus (SLE) by binding to a collagen-like region of C1q, thereby leading to C1 activation. Likewise, we can speculate that the C1q autoantibody plays a similar role in HUVS. However, C1q autoantibody was not detected in our patient.
Both HUVS and SLE are immune complex diseases that are associated with ANA, C1q antibody, and decreased complement. Previously, Her et al. (7) reported a Korean patient whose urticarial vasculitis preceded the manifestation of SLE, suggesting that HUVS could develop in a subset of SLE patients. However, both conditions are different in the main features of their cutaneous manifestation and overall pattern of systemic involvement. Namely, malar, discoid, or photosensitive rash are the predominant cutaneous manifestation in SLE unlike urticaria or angioedema in HUVS (8). In addition, chronic obstructive pulmonary disease and uveitis are typically not observed in SLE whereas those are complications seen in some HUVS patients (9). Our case fulfilled only 2 components of the Systemic Lupus International Collaborating Clinics (SLICC) criteria (10), i.e., renal involvement and low complement levels, insufficient for making a diagnosis of SLE. Moreover, his kidney biopsy did not show a ‘full house’ immunofluorescence pattern that is characteristic of lupus nephritis. Thus, we speculate that both conditions are different entities which share a similar pathogenesis and a subset of each one.
The renal involvement in HUVS occurs in about 14% of cases (6) with presentation of proteinuria or microscopic hematuria. Its development is believed to be the consequence of immune complex deposition (11), either of in situ formation of immune deposits or circulating immune complexes. Although it is known that renal disease is more severe in pediatric cases of HUVS, the clinical course of renal involvement in our patient was benign as indicated by the non-nephrotic range proteinuria and its complete remission without a decrease of the estimated glomerular filtration rate.
In regards to muscle involvement, there are a few examples in previous case reports (12). In our patient, recurrent proximal weakness and myalgia usually occurred within the same period as an episode of cutaneous symptoms and responded to glucocorticoid-based regimen, along with a reduction in CK and resolution of the skin lesions.
The optimal therapeutic approaches are yet to be established. The several changes to the regimen in our patient indicate that no particular combination fully controlled disease activity. The subsequent development of membranous nephropathy emphasizes the importance of regular monitoring for the progression of HUVS, even if the initial presentation does not manifest with systemic involvement or a significant decrease in the complement level.
 XML Download
XML Download