

 PDF
PDF Citation
Citation Print
Print


Abbreviations
CRS
chronic rhinosinusitis
CRSsNP
chronic rhinosinusitis without nasal polyps
CRSwNP
chronic rhinosinusitis with nasal polyps
CRTH2
chemoattractant receptor homologous molecule expressed on Th2 cells
CysLT
cysteinyl leukotriene
ILC
innate lymphoid cell
NP
nasal polyp
OSM
oncostatin M
PGD2
prostaglandin D2
PGP
Pro-Gly-Pro
STAT
signal transducer and activator of transcription
Th
T helper
TSLP
thymic stromal lymphopoietin
INTRODUCTION
Chronic rhinosinusitis (CRS) is chronic inflammation of the sinonasal mucosa and characterized by nasal congestion, rhinorrhea, and a decreased sense of smell persisting for greater than 12 weeks (1). CRS can be subdivided into two major categories based on whether nasal polyps (NPs) are present (chronic rhinosinusitis with nasal polyps [CRSwNP]) or absent (chronic rhinosinusitis without nasal polyps [CRSsNP]). CRS is a common inflammatory disease in paranasal sinus mucosa, affecting more about 10% of the Asian population (234). At least 10% of patients with CRS do not respond to medical and surgical treatment regimens and develop a refractory disease. Patients with CRSwNP often have higher disease severity and a risk of recurrence than do patients with CRSsNP (5) and various aspects of cellular responses were investigated to elucidate the immunological phenotypes of CRSwNP.
Phenotypes of CRSwNP, clinically observable characteristics with the presence of NP in the nasal cavity, have been classified merely based on histologic cellular infiltration in NP into eosinophilic or non-eosinophilic NP (6). The degrees of mucosal eosinophil infiltration have repeatedly been reported as a potent predictor of the poor outcome of surgeries and a risk of having comorbidities (7). However, there are no standards to evaluate tissue eosinophilia due to uneven distribution throughout the tissue, and all the reports are using their own methods to classify NP into eosinophilic or non-eosinophilic (8). There are quite significant racial and regional differences in the eosinophilic polyp proportion. Moreover, besides the eosinophils, various kinds and degree of inflammatory cells infiltrate in most of the NP, but the function and pathological roles of these cells are not yet completely elucidated (9). In even eosinophilic NP, neutrophils and macrophages exist and may have a role in nasal polypogenesis (3). Therefore, the current subtyping of CRS or NP based on inflammatory cells are too simple to explain the pathogenesis of CRSwNP. Due to these challenges, it is difficult to reach favorable treatment results and needs more research on other immune cells infiltrated in NP. CRS involves multiple pathogenesis mechanisms showing high heterogeneity, and those are suggested to cause different therapeutic responses. Characterization of endotypes of NP also helps to determine primary optimal therapeutic modality, select a good responder to a specific treatment and predict treatment outcomes and risks for comorbidities.
Though aberrant mucosal immune response in CRSwNP is observed and reported in the literature at the various aspects of view, it is unclear whether these phenomena are the cause or result of eosinophilic or neutrophilic inflammation. Furthermore, the primary mechanism of the recalcitrant or treatment-nonresponsive CRSwNP is not known. Moreover, in contrast to NP from Caucasian patients, NP from Asian patients are characterized by a much lesser portion of eosinophilic inflammation, suggesting other cells than eosinophils can play more important roles in CRS pathogenesis. Neutrophils and macrophages are more frequently observed in these population and expected to have potential roles in pathogenesis (1011). In this article, the recent research results in possible roles of innate immune cells are reviewed, and clues for the key mechanisms of recalcitrant CRSwNP were summarized.
EPITHELIAL CELLS
Epithelial-derived innate cytokines such as IL-25 and IL-33 may also participate in the evolution of NP (12). These cytokines are reported to exaggerate T helper (Th) 2 type immune responses in the typical Th2-prone diseases such as atopic dermatitis and bronchial asthma (1314151617). IL-33 is secreted by immune cells such as macrophages and dendritic cells as well as epithelial cells. IL-33 is constitutively expressed in epithelial barrier tissues including lung, skin, stomach and salivary gland in steady-state (1819). IL-33 expression can be further enhanced during inflammation. In allergic lower airway inflammation, IL-33 production is increased in the airway epithelial cells in asthma and as chronic obstructive pulmonary disease (COPD) (2021). The expression of IL-33 has also been observed in hepatocytes during acute hepatitis and in endothelial cells from the colitis tissue, in macrophages and dendritic cells during allergic inflammation and infection (222324). However, epithelial cells appear to be the most important major source of IL-33 as the level is suspected much higher in epithelial cells (2123).
Biologically active full-length IL-33 plays a role in mucosal inflammation recruiting neutrophils via chemokines including C-X-C motif chemokine ligand (CXCL)-1 and CXCL-2 (252627). Several studies sought to investigate the expression and role of IL-33 in CRS. There have been conflicting results on the expression of IL-33 in CRS. It was reported that IL-33 mRNA was highly expressed in nasal mucosa but was not elevated in NP or other inflamed areas of the sinuses in CRSwNP (282930). In vitro cell culture study, epithelial cell culture from NP, IL-33 mRNA levels were 3-fold higher in the recalcitrant group than the responsive group, and it was far more enhanced by CpG stimulation (31). A significant upregulation of ST2, a receptor for IL-33 was demonstrated in ethmoid mucosa from CRSwNP, but the concentration of IL-33 protein was not significantly different between NP and control tissue (32). Of note, the recent research conducted by our group demonstrated IL-33 was upregulated in neutrophil-dominant subtypes of CRSwNP compared to eosinophilic NP or controls in the Korean population (33). Accordingly, IL-33 level in tissue homogenate was correlated with Th1/Th17 inflammatory cytokines (33). However, in this study, protein levels of IL-5 and quantities of eosinophil infiltration were inversely related with levels of IL-33 which was shown in the previous studies conducted in the countries of eosinophil-dominant NPs, suggesting the difference of IL-33 role and immune mechanisms in subtypes of CRSwNP that shows regional differences.
IL-25 has been demonstrated to induce Th2 type immune responses. IL-25 is produced by eosinophils, mast cells, and airway epithelial cells (3435) and stimulates Th2 airway inflammation characterized by airway hyperreactivity, mucus production, airway eosinophilia, and increased serum IgE (3637). A recent investigation reported increased expression of IL-25 and its receptor in the airways of asthmatics after allergen provocation (38).
In CRS, some groups reported IL-25mRNA levels in CRSwNP, which was controversial. Our recent study showed IL-25 expression was upregulated at the protein level in the tissue of CRSwNP homogenate compared to normal healthy controls. These controversies are needed to be more investigated, however, given that Th1/Th2/Th17 inflammatory markers were positively correlated with IL-25mRNA level in Asian NP, the subtypes and regional or racial differences and severity of disease should be further elucidated in the future.
INNATE LYMPHOID CELLS (ILCs)
ILCs are RAG–independent cells with the absence of lineage markers for T/B cells and for other innate cells with lymphoid morphology (39). Three ILC groups have been identified in both mouse and humans, related to their dependence on the transcriptional repressor inhibitor of DNA binding 2 (Id2) and the IL-2Rγ chain (40). ILC2 is characterized by the capacity to produce large amounts of IL-5 and IL-13 when activated by the pro-Th2 cytokines IL-25, IL-33 or thymic stromal lymphopoietin (TSLP). Unlike T cells, these responses do not require antigen for activation, and they rapidly mobilize these cytokines, as named with ‘innate.’ ILC2s are regarded as innate counterparts of Th2 cells sharing same functional module by their mutual production of signature cytokines such as IL-5 and IL-13 (41).
About the role of ILCs in the pathogenesis of CRS, there are a vast amount of researches that are going on. ILC2s are most thoroughly studied in the context of lung immune cell homeostasis, immunity, and inflammation. In recent report, ILC2 are the central players in CRS pathogenesis. As ILC2s have been identified as a major innate producer of IL-13 and IL-5, in response to IL-25 and IL-33 expression (4243), its role in NP pathogenesis has been suggested.
In CRSwNP mucosa, it has been reported chemoattractant receptor homologous molecule expressed on Th2 cells (CRTH2)+ST2+ ILC2s are increased (44). TSLP activates human ILC2 by directly upregulating GATA3 and signal transducer and activator of transcription (STAT) 5, resulting in the production of high amounts of type 2 cytokines (4445). This observation is highly relevant in the context of CRS and also asthma, because TSLP protein expression was significantly increased in epithelial cells derived from NP of CRS patients and in the airway epithelium and lamina propria of asthmatic patients, particularly in patients with severe forms of diseases (46).
IL-33- and IL-25-activated ILC2s can induce eosinophilic airway inflammation accompanied by airway hyper-responsiveness even in Rag knockout mice, which means ILC2s function independent of acquired immunity (4748). ILC2s are abundant and also have a close relationship with higher tissue and blood eosinophilia in NP, clinically related to worsening nasal symptom scores and asthma comorbidity (4950). A recent study reported that there was spatial co-localization between ILC2s and eosinophils in NP. A co-culture of eosinophils and ILC2s augmented the activation of eosinophils and prolonged their survival, and in return, pre-activated eosinophils enhanced IL-5 production of ILC2s in an IL-4 dependent manner.
CRTH2 is the one of marker for human ILC2 (39). CRTH2 is known as the prostaglandin D2 (PGD2) receptor 2, binds PGD2 to induce eosinophilic Th2 inflammation, including ILC2 chemotaxis (515253). PGD2 induces the production of type 2 cytokines, IL-4, IL-5, and IL-13 in ILC2 by the activation of CRTH2. Mast cells are the most important source of PGD2 in NPs. Activated mast cell supernatant induced migration of ILC2 and Th2 cytokine production in ILC2 through CRTH2 (52). Mast cells produce other pro-inflammatory arachidonic acid metabolites cysteinyl leukotrienes (CysLTs) including LTC4 and LTD4 (54). ILC2 also expresses CysLT receptor 1, and CysLTs strongly induced type 2 cytokines in ILC2s (55). Given that mast cells were increased and activated in CRSwNP, and often found activated and degranulated in eosinophilic NPs (56), mast cells have potential to enhance ILC2 to induce type 2 immune responses in NPs.
It has been reported that IL-25 induces corticosteroid resistance through the induction of ILC2 in allergic pulmonary inflammation model (57). IL-25 induced-ILC2 transferring was sufficient to induce pulmonary allergic inflammatory responses.
In allergic asthma model induced by allergen co-administered with IL-33, the ILC2-related TSLP-STAT5 pathway has been suggested to be critical for steroid resistance to airway inflammatory disease. Steroid resistance of ILC2s was recently reported as TSLP dependent (58), where IL-33 in combination with antigen resulted in ILC2s with increased resistance to steroid treatment than T cells. This process required STAT5 and could be inhibited with pimozide (a STAT5-inhibiting antipsychotic drug). Treatment-recalcitrant ILC2 may contribute to disease heterogeneity which might lead to recurrence and exacerbation of CRSwNP.
The role of ILCs in non-eosinophilic CRSwNP was not clear, and there are very few report that ILC3s are more frequently found in CRSwNP. In the recent study, the role of ILC3 in obesity-associated asthma was reported. Airway hyperreactivity in obesity-related asthma mouse model was dependent on IL-17A and the NLRP3 inflammasome, and the possible link with ILC3 was demonstrated that this phenotype could be adoptive transferred by IL-17 producing ILCs. Though the role of ILC3 in NP pathogenesis is not clear, this suggests that ILC3s have the potentials in non-eosinophilic NP pathogenesis, which needs further investigations (59).
MACROPHAGE
Macrophages are generated in the bone marrow by progenitor cells. The life-span of macrophage is varying from hours to possibly years depending on the type of immune response. Tissue macrophages are differentiated from circulating monocytes when they enter tissue. Tissue macrophages have a role functioning as the phagocyte ingesting pathogens, a scavenger of dead cells and debris, and a leading player of tissue remodeling after damage (6061).
Based on their function, macrophages are subdivided into M1 macrophage, mediating host defense and anti-tumor immunity, and M2 macrophage, suppressing immune response with regulating wound healing and some other macrophages that have immunosuppressive activity, regulatory macrophage that secrete IL-10, tumor-associated macrophages that suppressed anti-tumor immune response, and the monocytic subset of myeloid-derived suppressor cells. Macrophages can have important roles in both immune induction and resolution (62).
Focusing on the chronological order of CRSwNP, comparing mature ethmoidal polyps or normal mucosa, subepithelial eosinophils and M2 type macrophages were significantly increased in numbers in the early stage polyps confined to middle turbinate (63).
In a recent report, it was demonstrated that group V phospholipase A2 and transglutaminase-2 was co-localized in macrophages of human NP tissue obtained from patients with Th2 type eosinophilic inflammation, and their co-expression positively correlated with the number of eosinophils in each tissue specimen (64). In human M2 macrophage activated with IL-4, group V phospholipase A2 PLA2G5 regulates transglutaminase activity of human IL-4-activated M2 macrophages through PGE2 generation. Group V phospholipase A2 is a functionally relevant enzyme that may have therapeutic value for the treatment of human Th2 inflammatory CRSwNP.
The number of CD68+ CD163+ alternatively activated (M2) macrophages was increased in eosinophilic NP. In eosinophilic Th2 milieu, M2 macrophage expresses tumor necrosis factor-α-induced protein 8-like 2 (TIPE2), a new candidate of immune modulating protein, by IL-4 and IL-13 stimulation. TIPE2+ CD163+ CD68+ M2 macrophage correlated with eosinophilic inflammation and clinical refractoriness (65). In tissue remodeling by Th2-induced coagulopathy, M2 macrophages, alternatively activated by Th2 cytokines (66), produce coagulation factor XIIIa which acts enzymatically and contribute to forming a tight tetrameric complex (FXIIIA2B2), cross-linking process of fibrin, culminating in edematous remodeling patterns in NP (67).
NEUTROPHIL
Neutrophils have long been described in the phenotype studies of CRSwNP. Neutrophil-dominant CRSwNP were reported mainly in Asian NP. CRSwNP was initially classified according to the presence of NP which reflects eosinophils-infiltrating Th2 inflammation predominantly at the molecular levels in Western countries (68). However, Asian populations such as in China showed multiple inflammatory cells infiltrate, including neutrophils and T cells classified as Th1/Th17 cells (10). Furthermore, mixed Th17/Th2 inflammatory patterns were also demonstrated throughout a single NP tissue, and even eosinophilic NPs encompass neutrophils and expresses its biological markers such as IL-8 and myeloperoxidase (3).
Similar to M1 and M2 in subpopulations of macrophages, neutrophils are grouped into either N1 or N2. Like the functions in the subpopulation of M1 and M2 macrophage, N1 and N2 neutrophils are functioning as N1 to be pro-inflammatory, whereas the N2 as an anti-inflammatory. N1 neutrophils have been shown to express more pro-inflammatory cytokines and chemokines, such as tumor necrosis factor (TNF), IL-1β, C-C motif chemokine ligand (CCL) 3, CCL5, IL-6, and IL-12. Compared to N1 neutrophil, N2 neutrophils are the immunosuppressive counterpart. N2 neutrophils are related to tumor progression, metastasis, and like M2 macrophage, are the players in tissue repair and inflammation-resolving process. Tumor-associated N2 neutrophils are characterized by high expression of C-X-C chemokine receptor type 4 (CXCR4), vascular endothelial growth factor and matrix metalloproteinase-9 and are induced in the presence of high levels of transforming growth factor-β (TGF-β).
The polarization and markers for N1 and N2 were more studied in mice. Till now, there are no clear-cut phenotypical markers for neutrophilic subsets, however, based on several studies mainly in mice, have shown part of results using similar markers that distinguish M2 macrophages (6970). It will be similar to M2 macrophage: express the mannose receptor C-type 1 (MRC1), arginase 1 (ARG1), chitinase-like 3 (Ym1), IL-10, and TGF-β.
Recently the pathogenic role of neutrophils in CRSwNP was reported disrupting epithelial barrier by producing oncostatin M (OSM) (7172). These studies demonstrated OSM-producing neutrophil by flow cytometry analysis and immunofluorescence imaging. This neutrophil population produces OSM by the stimulation with granulocyte-macrophage colony stimulating factor (GM-CSF), however, not by the stimulation with IL-25, IL-33, or TSLP. It was suggested neutrophil itself could produce GM-CSF, thus there exists autocrine pathway to secrete OSM in NP. These OSM-producing neutrophils from NP expressed the Th2 type immune response marker ARG1 analyzed by FACS, indicating a phenotype resembling N2 neutrophils more than N1 neutrophils.
Neutrophils are traditionally regarded as the cell of acute inflammation with relatively short life span in tissue, and the reason and the role of accumulation of neutrophil in CRSwNP pathogenesis are not known (73). The similar chronic neutrophilic inflammation is observed in lung diseases such COPD or cystic fibrosis (CF). The mechanism of prolonged excessive neutrophil accumulation is suspected to be driven by tripeptide N-acetyl Pro-Gly-Pro (PGP), the product derived from the breakdown of the extracellular matrix of lung tissue in the disease state (74). Neutrophil product secreted in the inflammatory process may attribute this process, including various proteolytic enzymes that may alter the protease-antiprotease balance. Additionally, cigarette smoking selectively inhibits leukotriene A4 hydrolase aminopeptidase activity, which naturally degrades PGP and resolving inflammation, resulting in the accumulation of PGP and neutrophils (75). However, whether this process is taken place in neutrophil-dominant CRSwNP is largely unknown.
Neutrophils are not classically associated with Th2 immune responses and are considered a Th1 immune response effector cell. However, although the previous study has been conducted with NP in Western countries which tend to be highly eosinophilic, they had significant numbers of neutrophils (71). In parallel, increased neutrophilia within NP has been linked to decreased responsiveness to corticosteroid treatment and neutrophilic subsets of NP are also reported especially in Asian polyps (7677), neutrophils may contribute to the pathogenesis and treatment resistance of NPs even though little is known.
CONCLUSION
This article focused on the review of the recent research about key candidate innate immune player cells in the pathogenesis of CRSwNP. The pathogenesis of NP described above is summarized in Fig. 1.
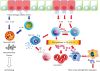
Targeting acquired immunity and mainstay of research targeting Th2 inflammation has been progressed actively, launching the new treatment targets such as monoclonal antibodies against IL-5, IL-4 receptor alpha subunit, IgE, and TSLP in recent clinical trials. The therapies targeting Th2 inflammation showed the beneficial effect especially on eosinophilic NP and/or asthma.
Although the majority of CRSwNP cases in western countries show type 2 inflammation and eosinophilia, non-eosinophilic CRSwNP is more common in Asian countries (39). Given that a worse prognosis for steroid therapy in non-eosinophilic CRSwNP versus eosinophilic CRSwNP was demonstated (76), more studies on innate inflammatory molecules in non-eosinophilic CRSwNP can develop novel therapeutic targets for recalcitrant CRSwNP. CRSwNP is a heterogeneous disease and more precise sub-classification concerning the inflammatory mechanism reflecting prognosis and predicting target treatment modality of this disease might be required in the future.
 XML Download
XML Download