

 PDF
PDF Citation
Citation Print
Print


Abbreviations
AS
ankylosing spondylitis
COPD
chronic obstructive pulmonary disease
DKK-1
dickkopf-1
FLS
fibroblast-like synoviocytes
GPA
granulomatosis with polyangiitis
IFN
interferon
OB
osteoblast
OC
osteoclast
PBMC
peripheral blood mononuclear cell
PR3
proteinase 3
RA
rheumatoid arthritis
RANKL
receptor activator of NF-κB ligand
SLE
systemic lupus erythematosus
TG
transgenic
TNF
as tumor necrosis factor
WT
wild-type
INTRODUCTION
Chronic inflammatory conditions such as rheumatoid arthritis (RA) and ankylosing spondylitis (AS) disrupt the balance of bone homeostasis by accelerating the formation of osteoclasts (OCs) or osteoblasts (OBs) (12). Bone destruction in RA is caused by a combination of inflammatory cytokines, such as tumor necrosis factor (TNF)-α, IL-6, and IL-17, in the affected joints, which induce differentiation and activation of OCs (1). Although TNF-α and IL-17 are pivotal cytokines that regulate inflammation in AS, the main feature of joints in AS are bony ankyloses characterized by excessive OB activation leading to the formation of syndesmophytes (2). Considering the differences between RA and AS, a novel cytokine associated with pathological bone metabolism may exist.
IL-32, also known as natural killer cell protein 4 (NK4), has been defined as an inflammatory cytokine involved in infection, cancer, chronic inflammation, and autoimmune diseases (34567). In response to various inflammatory stimuli, IL-32 is produced by both immune and non-immune cells, including NK cells, T cells, monocytes, epithelial cells, endothelial cells, and fibroblasts (8910). During infection, IL-32 activates inducible nitric oxide synthase, interferon (IFN)-λ1, and IL-6, resulting in antiviral effects (11). Further, IL-32γ transgenic (TG) mice have shown resistance to lipopolysaccharide-mediated septic shock by reducing systemic cytokine release, indicating a protective effect against bacterial infection (12). IL-32 expression is high in a variety of cancers, including gastric cancer, lung cancer, hepatocellular carcinoma, and pancreatic cancer (13141516). IL-32 promotes the growth, invasion, and metastasis of tumors (1314).
In humans, there are at least 9 different isoforms of IL-32 (IL-32α, IL-32β, IL-32γ, IL-32δ, IL-32ε, IL-32ξ, IL-32η, IL-32θ, and IL-32ζ) generated by alternative splicing (17). IL-32γ is the longest (full protein) and most active isoform in terms of stimulating peripheral blood mononuclear cells (PBMCs) or producing pro-inflammatory cytokines in macrophages (18). IL-32α, which is the most abundant isoform, is the shortest and the least active pro-inflammatory isoform (18). The detailed functions of these individual isoforms and their corresponding receptors, as well as why there are up to 9 different isoforms, remain unclear (19). However, a previous study suggested that splicing of IL-32γ to shorter isoforms represents a safety switch mechanism to protect against uncontrolled and exaggerated inflammation (20).
Among the isoforms of IL-32, IL-32γ has been shown to be involved in the pathogenesis of RA and AS, both of which are inflammatory joint diseases (212223). In this review, we describe the multifaceted role of IL-32γ, particularly in bone-related phenotypes in chronic inflammatory autoimmune arthritis, and explain the distinct clinical aspects of RA and AS involving this cytokine.
ROLE OF IL-32 IN INFLAMMATORY DISEASES
IL-32 has diverse roles in mediating chronic inflammation and autoimmunity (6). An elevated concentration of serum IL-32 has been observed in patients with granulomatosis with polyangiitis (GPA) (24) and those with chronic obstructive pulmonary disease (COPD) (25). In GPA, the IL-32 concentration was elevated and altered according to the treatment response. Neutrophil proteinase 3 (PR3) is a major autoantigen of GPA (24), and IL-32α binds to PR3 with high affinity (26). By inducing proinflammatory cytokines such as TNF-α and IL-6 and binding to PR3, IL-32 appears to be closely associated with responsiveness to treatment in patients with GPA (24). In COPD, increased serum concentrations of IL-32 correlates with smoking status (25). Inflammation in COPD is induced by type 1 helper T cells and the production of cytokines, such as IFN-γ (2728). IFN-γ is one stimulator of IL-32 production (8). Thus, an increased serum concentration of IL-32 in patients with COPD can be explained by the increased levels of IFN-γ (25). Moreover, expression of IL-32 in the lung tissue of patients with COPD has been shown to be negatively correlated with lung function parameters and positively correlated with TNF-α expression, number of CD8+ cells infiltrating lung tissue, and phosphorylation state of p38 mitogen-activated protein (9). Thus, IL-32 has been implicated in COPD immune response and disease progression (9).
IL-32 is also associated with the progression and pathophysiology of inflammatory bowel diseases (629). The secretion of IL-32 is enhanced by the intracellular nucleotide oligomerization domains and muramyl dipeptide, which is a peptidoglycan fragment from bacteria and potent nucleotide oligomerization domain 2 ligand that induces the expression of IL-32 in a caspase-1-dependent manner (30). This leads to increased production of IL-6 and IL-1β (3031). IL-32ε, an isoform of IL-32, has been identified in human colonic subepithelial myofibroblasts and found to be enhanced in the inflamed mucosa of patients with inflammatory bowel disease (32). The role of IL-32 in bowel inflammation has been investigated in vivo using IL-32γ TG mice (333), in which an increased concentration of IL-32γ was associated with faster and more severe progression of acute bowel inflammation compared to wild-type (WT) mice. However, the degree of colonic inflammation was lower, and the survival rate was higher, with dextran sodium sulfate-induced colitis in IL-32γ TG mice (333). The lower degree of inflammation can be explained by the splicing of IL-32γ into IL-32β, which may induce IL-10 and lead to an increased anti-inflammatory response (34).
In systemic lupus erythematosus (SLE), the importance of IL-32 remains controversial. One study found no significant difference in the serum concentrations of IL-32 between SLE patients and healthy controls (35), whereas another study reported a high concentration of IL-32γ in some patients with SLE with lupus nephritis (36). Despite anecdotal reports showing increased serum concentrations of IL-32γ in SLE, the functional role of IL-32γ in SLE pathogenesis is unclear. Additionally, it remains unknown whether the serum concentrations of other IL-32 isoforms are elevated in SLE. Further studies are required to confirm the roles of IL-32γ and other isoforms in patients with SLE.
In a study of skin disorders, an association between IL-32 and atopic dermatitis has been reported. One study showed that human keratinocytes express high levels of IL-32 under stimulation by IFN-γ, TNF-α, and type 1 helper T cells (37). Further, transfection of keratinocytes with small interfering RNA to IL-32 significantly reduced keratinocyte apoptosis, suggesting that IL-32 stimulates apoptosis of keratinocytes. Expression of IL-32 was increased in skin biopsy specimens from patients with atopic dermatitis compared to in those from healthy donors or patients with psoriasis, showing that IL-32 contributes to the development of atopic dermatitis by stimulating the apoptosis of keratinocytes.
The association of IL-32 with disease activity is also evident in RA, as shown by the positive correlation of IL-32γ with TNF-α in the peripheral blood of active patients with RA (38). TNF-α and IL-1β stimulate the production of IL-32γ from RA fibroblast-like synoviocytes (FLS) (38), and the concentration of IL-32γ has been shown to be elevated in the joint fluid of patients with RA (23). Additionally, joint swelling, inflammatory cell infiltration, and cartilage damage can be induced by injecting IL-32γ into mice knee joints (39), and severe synovitis and cartilage damage can be induced in IL-32α TG mice (and not WT mice) with lipopolysaccharide knee injections (40).
A recent report showed that a high concentration of IL-32γ accumulated in the joint of patients with AS and IL-32γ was expressed in the synovia of AS patients at a much higher concentration than in the synovia of RA patients (22). However, the concentration of IL-32γ in the peripheral joints was not significantly correlated with systemic inflammation and AS activity indices (22).
ROLE OF IL-32 IN BONE METABOLISM
OC and bone resorption by IL-32γ
In RA, which is characterized by chronic synovitis and hyperplasia, the joints are the representative pathological sites of progressive cartilage and bone destruction (41). Activated macrophages are the predominant cell type in the inflamed synovial tissue and are responsible for promoting inflammation via the production of inflammatory cytokines, such as TNF-α, IL-17, IL-1β, and IL-6 (41). These cytokines promote the differentiation of preosteoclasts into mature OCs, enhance bone resorption, and prolong OC survival, thereby contributing to bony erosion in inflamed joints (42). Receptor activator of NF-κB ligand (RANKL), which binds to RANK, is a key molecule in the differentiation of OCs (43). RANKL is a membrane-bound homotrimeric protein found in the OB lineage, and its soluble form (sRANKL) is abundantly produced from FLS in conditions associated with joint inflammation, such as RA (44). TNF-α induces RANKL and RANK expression and thereby stimulates OC differentiation via RANKL-induced osteoclastogenesis (4546). Additionally, TNF-α can directly induce OC differentiation in the absence of RANKL by activating NF-κB signaling (47). IL-17 stimulates RANKL expression and thus RANKL-induced osteoclastogenesis (48). However, in the absence of RANKL, IL-17 can also induce OC differentiation by enhancing TNF-α-induced osteoclastogenesis (49).
Expression of IL-32 in synovial tissue has been extensively studied in patients with RA (233950). Joosten et al. (39) reported that IL-32 was elevated in RA synovial tissue compared to tissues from patients with osteoarthritis and healthy controls, and that the expression of IL-32 in synovial staining correlated with synovial inflammation and the serum erythrocyte sedimentation rate. Furthermore, in a comparison of joint inflammation in WT mice versus TNF-α gene knockout mice after IL-32γ injection into the joint, TNF-α gene knockout mice showed no evidence of joint swelling and had reduced numbers of inflammatory cells in their synovial tissue compared to WT mice (39). This shows that IL-32γ-mediated joint inflammation in RA synovial tissues is at least in part TNF-α-dependent (39). The relationship between IL-32β and TNF-α in vivo was studied by Shoda et al. (10) using a collagen-induced arthritis model. Transfer of IL-32β-producing CD4+ T cells to collagen-immunized mice exacerbated collagen-induced arthritis, with TNF-α blockade attenuating this exacerbation, revealing a close association between IL-32β and TNF-α (10). Furthermore, RA-derived FLSs produce IL-32 in response to TNF-α in a dose-dependent manner, suggesting that the inflammatory cascade in the synovial tissue of RA can be amplified by IL-32 activity via an autocrine loop (50).
The first study of IL-32 and OCs was carried out using IL-32α by Mabilleau and Sabokbar (51); they investigated the effect of IL-32α on OC differentiation and activation. OC differentiation is a process in which OC precursors develop into mature OCs, which express genes that typify the OC lineage (NFAT c1 [NFATc1], TNF receptor-associated factor 6, OC-associated receptor and cathepsin K) (52). In contrast, OC activation is a process in which mature OCs are activated, resulting in the initiation of bone resorption (52). Mabilleau and Sabokbar (51) reported that IL-32α induced the differentiation of OCs but did not activate these multinucleated cells into bone-resorbing OCs. However, IL-32γ, the isoform with a more potent biological mechanism for stimulating PBMCs than IL-32α (18), stimulates and activates OC differentiation, which can be confirmed in a bone resorption assay. This protein is activated via NFATc1 activity and exerts synergistic effects with RANKL on osteoclastogenesis (23). According to a previous study (18), the biological activities in TNF-α and IL-6 secretion from PBMCs differed between the IL-32α and IL-32γ, suggesting that the different biological activities of the 2 isoforms were attributed to differences in OC activity.
Interestingly, the synergistic effect of RANKL-induced signaling and IL-32γ on osteoclastogenesis was demonstrated by treating cells with IL-32γ at the fusion stage (53). Kim et al. (23) demonstrated that IL-32γ induced the expression of dendritic cell-specific transmembrane protein, which suggests that IL-32γ is a mediator of OC fusion. Furthermore, IL-32γ can induce the differentiation of CD14+ monocytes into OCs even in the absence of sRANKL stimulation; however, bone-resorbing activity is not sufficient in RANKL-independent IL-32γ-induced OCs.
In contrast, IL-32γ suppresses the transcription of osteoprotegerin in RA-FLS and OBs (2123). Collectively, IL-32γ forms an osteoclastogenic environment in patients with RA by promoting OC differentiation, exhibiting synergistic effects with RANKL, and suppressing osteoprotegerin (23). Thus, inhibition of IL-32γ under RANKL-rich conditions including RA may be useful for delaying or preventing inflammation or tissue destruction.
OB and bone formation by IL-32γ
AS is a chronic inflammatory form of arthritis that primarily affects the spine and large joints. New bone formation in AS may be related to an active repair process following damage caused by inflammation (5455), which is bone remodeling characterized by sequential and local communication between OBs and OCs (56). Previous reports showed that IL-32γ stimulates OC formation in vitro (7235758) while actively enhancing OB differentiation (2122), indicating a physiological role for IL-32γ in the bone remodeling process.
Lee et al. (22) first reported the pathogenic role of IL-32γ in AS and OB differentiation using IL-32γ TG mice. Although the TNF-α concentration was not significantly different between AS and RA joint fluids, IL-32γ accumulated in the inflamed joints of patients with AS at a much higher degree than in patients with RA. Additionally, immunohistochemical staining revealed high expression of IL-32γ in the paravertebral soft tissues and peripheral synovia in patients with AS. Moreover, IL-32γ TG mice showed a higher degree of osteogenic differentiation compared to WT mice and administration of IL-32γ induced OB differentiation actively in vitro (2122). These data suggest that IL-32γ is involved in AS pathogenesis, particularly in bone progression.
Dickkopf-1 (DKK-1) is a potent Wnt pathway inhibitor produced by OBs and is known to suppress OB differentiation. Serum DKK-1 concentrations are inversely correlated with spinal bone progression in patients with AS (59), and blockade of DKK-1 promotes the formation of ankylosis of sacroiliac joints in model mice (60). Recently, Lee et al. (21) demonstrated a relationship between IL-32γ and DKK-1 in bone metabolism and the functional mechanism of osteogenesis mediated by systemic IL-32γ. In their study, human IL-32γ TG mice showed increased bone formation and reduced trabecular bone loss induced by ovariectomy (21). To clarify the molecular mechanism of IL-32γ-mediated downregulation of DKK-1, the authors also investigated the differential regulation of microRNAs by IL-32γ. Interestingly, miR-29a in primary OBs from IL-32γ TG mice was expressed at a significantly higher level than in the WT group (21). However, despite the protective function of IL-32γ in bone loss demonstrated in the study, the regulatory mechanism of IL-32γ alteration — except for the role of miR-29a — was not conclusively determined.
Furthermore, a study found that among osteoporosis patients, hip fracture victims had lower IL-32γ concentrations in their blood than those who did not experience hip fractures (21). The same study found that patients with factured hips had higher DKK-1 blood concentrations than those with intact hips. Interestingly, there was no significant difference in the bone marrow concentrations of IL-32γ between osteoporosis patients with and without hip fractures, indicating that reduction of systemic IL-32γ rather than local IL-32γ is responsible for osteoporotic fractures (21). Thus, in patients with osteoporosis, a combination of low IL-32γ and high DKK-1 concentration in the blood can be used as a predictive marker for osteoporotic fracture.
Collectively, IL-32γ may act as a key regulator in the pathogenesis of AS by controlling DKK-1 expression, leading to modulation of Wnt/β-catenin signaling. Therefore, IL-32γ may be a promising novel molecular target for preventing atypical bone formation in patients with AS (22).
CONCLUSION
IL-32γ is involved in bone changes associated with inflammatory arthritides, such as RA and AS. IL-32γ, through its synergistic effects with RANKL secreted from RA-FLSs and OBs, promotes OC formation and activation and contributes to inflammatory bone loss in RA. In contrast, locally elevated IL-32γ in AS may be associated with excessive bone formation by enhancing OB differentiation by inhibiting DKK-1 (Fig. 1). Thus, IL-32γ may be a promising biomarker of bone quality and a useful target for therapeutic applications in inflammatory bone diseases.
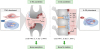 | Figure 1Multifaceted roles of IL-32γ in bone metabolism in RA and AS. RA is characterized by bone destruction, whereas AS is characterized by bone formation. In RA, IL-32γ and TNF-α promote OC formation by increasing RANKL production from RA-FLSs and OBs, thereby contributing to inflammatory bone loss in RA. However, in AS, highly elevated IL-32γ in the joint plays a key role in excessive bone formation by enhancing OB differentiation via inhibition of DKK-1.
|
 XML Download
XML Download