

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
The oral epithelium covers and protects the internal structures of the oral cavity from the external environment. Therefore, the oral epithelium forms a barrier between oral bacteria and the tissue that provides the first line of defense against invading bacteria. The physical barrier function of the epithelium mainly depends on tight junctions (TJs) and adherens junctions (AJs) [1]. The TJ proteins expressed in the oral epithelium include junctional adhesion molecule (JAM)-A, occludin, claudins, zonula occludens (ZO)-1, and ZO-2, while the AJs consist of E-cadherin, nectins, and β-catenin [2]. The expression levels and tight assembly of these junctional proteins are important for the maintenance of the barrier function [1].
Across various anatomical sites, epithelial barrier dysfunction is involved in the pathophysiology of diverse inflammatory diseases, such as atopic dermatitis, asthma, chronic rhinosinusitis, inflammatory bowel diseases, periodontitis, and oral lichen planus [345678]. It is well known that invading pathogens disrupt the epithelial barrier to gain access into the host tissue. For example, the periodontal pathogens Porphyromonas gingivalis, Treponema denticola, and Aggregatibacter actinomycetemcomitans release virulence factors, such as gingipain, dentilisin, and cytolethal distending toxin, respectively, that induce damage or remodeling of the TJs and AJs of gingival epithelial cells [91011]. Infiltrated immune cells also increase epithelial permeability by releasing inflammatory cytokines, such as tumor necrosis factor alpha (TNFα), interleukin-1β, and interferon-gamma, proteases, and reactive oxygen species [12].
In contrast, several growth factors or hormones, such as epidermal growth factor and estrogens, are known to reinforce epithelial barrier function [13]. Estrogens have diverse actions in non-reproductive systems, as well as in the reproductive system. There are 3 major forms of physiological estrogens: estrone, estradiol (E2), and estriol (E3). E2 is the most potent estrogen and the major product synthesized during the premenopausal period [14]. The functions of the estrogens are mainly mediated through 2 types of nuclear receptors, estrogen receptor (ER) α and ERβ, but also through membrane receptors such as ER-X and GPR30 or ER independently [14]. Estrogens enhance the physical barrier function of intestinal and esophageal epithelia through ERβ-mediated upregulation of TJ proteins [151617]. Expression of ERβ in the human oral epithelium of both genders has been reported [18]. However, the role of estrogens in the regulation of oral epithelial homeostasis has not been studied. Thus, the aim of this study was to investigate the effect of E2 on the physical barrier and regulation of TJ proteins in human oral epithelial cells.
MATERIALS AND METHODS
Human epithelial cell culture
Immortalized human oral keratinocyte (HOK-16B) cells originating from retromolar gingival tissues [19] were maintained in keratinocyte growth medium supplemented with supplementary growth factor bullet kit (Clonetics Corp., San Diego, CA, USA) in an atmosphere with 5% CO2 at 37°C.
Culture of epithelial cell monolayers and measurement of transepithelial electrical resistance (TER)
To investigate the effects of E2 and dexamethasone (Dexa) on the barrier formation in oral epithelial cells, 1×105 HOK-16B cells/well were seeded on transwells with a polycarbonate membrane with 3-μm pores and an area of 0.33 cm2. One day after seeding, the cells were treated with 0–20 nM E2 (Sigma-Aldrich, St. Louis, MO, USA) alone, 250 nM Dexa alone, or co-treated with E2 and Dexa, and the TER was measured at the indicated time points using an Electrical Resistance System Volt-Ohm Meter. The concentration of Dexa was determined by preliminary experiments.
To examine the effect of TNFα on the epithelial barrier, 1×105 HOK-16B cells/well were seeded on transwells and cultured for 2–3 days with daily medium changes until the confluent monolayers reached a peak resistance of approximately 14 Ω. The tight-junctioned monolayers of HOK-16B were treated with various concentrations of TNFα (R&D Systems, Minneapolis, MN, USA), and the TER was assessed at 0, 2, 4, 8, and 24 hours.
To determine the protective effect of E2 on the epithelial barrier from the TNFα-induced damage, the tight-junctioned monolayers of HOK-16B were pre-treated with 125 μM ICI 182,780 (Sigma-Aldrich) for 6 hours and/or 2-20 nM E2 for 4 hours. The cells were then treated with 100 ng/mL TNFα for 24 hours, and the TER was measured. All experiments were repeated twice in triplicate.
The cell counting kit (CCK)-8 assay
When the HOK-16B cell monolayers were cultured for the TER measurements, the cells were plated onto 96-well plates in parallel and cultured under the same conditions. Each plate was prepared for each time point. To measure cell proliferation and cytotoxicity, CCK-8 solution (Dojindo, Tokyo, Japan) was added to each well after removing the culture medium. The cells were further incubated for 1 hour, and the optical absorbance was then measured at 450 nM using a microplate reader (Molecular Devices, San Jose, CA, USA). The cell viability was calculated as the relative percentage of the vehicle control. All experiments were repeated twice in triplicate.
Immunofluorescence staining and confocal microscopy
To determine the effects of E2 on the expression levels of ZO-1 and JAM-A during barrier formation, 2×105 HOK-16B cells were plated onto 12-mm-diameter collagen-coated cover slips in a 24-well plate for 1 day and then treated with E2 for 24 hours. For the TNFα-induced damage condition, 5×105 HOK-16B cells were plated onto 12-mm-diameter collagen-coated cover slips in a 24-well plate and cultured for 2 days. The tight-junctioned monolayers of HOK-16B were incubated with 10 or 100 ng/mL TNFα for the indicated time in the absence or presence of E2 or ICI 182,780 (125 μM).
The cells were fixed with 4% paraformaldehyde and treated with 50 mM ammonium chloride for 10 minutes to quench autofluorescence. After permeabilization with 0.3% phosphate buffered saline with tween 20 and blocking with 5% bovine serum albumin, the cells were incubated with rabbit anti-ZO-1 polyclonal antibody (Invitrogen, Carlsbad, CA, USA), mouse anti-JAM-A antibody clone 2E3-1C8 (Abnova, Taipei, Taiwan), or anti-nuclear factor (NF)-κB p65 polyclonal antibody (Biolegend, San Diego, CA, USA) overnight at 4°C, and then with Alexa 488 anti-rabbit antibody, Alexa 555 anti-mouse antibody, or Alexa 555 anti-rabbit antibody for 1 hour. After washing with phosphate buffered saline, the cells were counterstained with Hochest 33342 and mounted. For each slide, 10 areas were photographed at ×100 magnification using a Zeiss LSM700 (Zeiss, Jena, Germany) with serial Z-sections. Maximum intensity projections were obtained by combining the serial Z-sections. The fluorescence intensity of ZO-1 and JAM-A was analyzed using ZEN 2010 (Zeiss) and normalized to the fluorescence intensity of Hochest 33342. For quantification of nuclear translocation of NF-κB, co-localized signals of the nucleus and NF-κB p65 were measured using the ImageJ software (National Institutes of Health, Bethesda, MD, USA; https://imagej.nih.gov). Experiments were repeated 2 or 3 times.
Statistics
All data are presented as mean±standard error of the mean. For the differences between the control and experimental groups, the 2-tailed non-paired Student's t-test was performed. One-way analysis of variance with the Tukey post hoc test was used to assess the differences among multiple groups. Significance was set at P<0.05.
RESULTS
E2 facilitates the formation of a physical barrier in oral epithelial cell monolayers
We first examined the effect of E2 on physical barrier formation by oral epithelial cell monolayers. HOK-16B cells plated onto the transwells were treated with various concentrations of E2, and the TER was measured at 0, 6, 12, 24, 48, 72, and 96 hours. Compared to baseline, the TER of the control cells treated with vehicle alone increased continuously until 96 hours, reaching a peak resistance of 14.4 Ω. Increases in the TER by E2 treatment were observed in a dose-dependent manner until 72 hours, although statistical significance was only achieved by the 20 nM concentration of E2 at 6 and 24 hours (Figure 1A). The increase in the TER by the E2 treatment was not due to an increased number of cells, because E2 did not affect cell proliferation, which was determined by the CCK-8 assay (Figure 1B).
 | Figure 1Facilitation of barrier formation by E2 in oral epithelial cell monolayers. (A) Immortalized human oral keratinocyte (HOK-16B) cells seeded on the membrane of a transwell 2-chamber tissue culture system were treated with various concentrations of E2 during barrier formation. The TER was measured at the indicated time points and expressed as percentage of the baseline (0 hours). (B) HOK-16B cells plated onto 96-well plates in parallel were treated and cultured in the same way. The proliferation of HOK-16B cells was measured using the CCK-8 assay kit at the indicated time points. The measured absorbances are expressed as the relative index compared to the vehicle control at each time point. (C) HOK-16B cells plated on collagen coated cover slips were treated with 2 or 20 nM E2 for 24 hours. After fixation, cells were stained for JAM-A and ZO-1 and examined by confocal microscopy with serial z-sections. Images with a maximal intensity that combined the serial z-sections are shown. (D) The fluorescence intensities of JAM-A and ZO-1 were analyzed and normalized to the fluorescence intensity of Hochest 33342.E2: estradiol, TER: transepithelial electrical resistance, CCK: cell counting kit, JAM: junctional adhesion molecule, ZO: zonula occludens, TJ: tight junction.
a)P<0.05 versus vehicle control (2-tailed non-paired Student's t-test).
|
To understand the mechanism for the increase in TER caused by E2, changes in the levels of TJ proteins were examined by confocal microscopy after treating the cells with E2 for 24 hours. E2 increased the levels of both JAM-A and ZO-1 in a dose-dependent manner, although statistical significance was only achieved by the 20 nM concentration of E2 (Figure 1C and D). These data suggest that E2 facilitates barrier formation through up-regulation of TJ proteins in human oral epithelial cells.
E2 protects against TNFα-induced damage in the barrier function of oral epithelial cell monolayers
The inflammatory cytokine TNFα has been shown to increase the permeability of human gingival epithelial cells [20]. The concentration of TNFα that impairs the barrier function of oral epithelial cells was determined by treating HOK-16B cell monolayers with 10–250 ng/mL TNFα after the TER reached its peak. All tested concentrations of TNFα decreased the TER in a time-dependent manner (Figure 2A); however, the 250 ng/mL concentration also showed significant cytotoxicity (Figure 2B). Treatment with TNFα decreased the levels of the ZO-1 and JAM-A proteins at 24 hours in a dose-dependent manner (Figure 2C and D). Based on these results, 100 ng/mL was chosen as the concentration of TNFα used in the subsequent experiments.
 | Figure 2Disruption of the physical barrier by TNFα. Tight-junctioned monolayers of human oral keratinocyte (HOK-16B) cells were treated with various concentrations of TNFα for 24 hours. (A) The TER was measured at the indicated time points. (B) Cell viability was measured using the CCK-8 assay kit at 24 hours. (C) After fixation, cells were stained for JAM-A and ZO-1 and examined by confocal microscopy. (D) The fluorescence intensities of JAM-A and ZO-1 were analyzed.TNFα: tumor necrosis factor alpha, TER: transepithelial electrical resistance, CCK: cell counting kit, JAM: junctional adhesion molecule, ZO: zonula occludens, TJ: tight junction.
a)P<0.05 versus 0 ng/mL (2-tailed non-paired Student's t-test).
|
To investigate the effect of E2 on the barrier function of oral epithelial cells under inflammatory conditions, HOK-16B cell monolayers were pre-treated with either E2 alone or E2 together with ICI 182,780, a nuclear ER antagonist, before treatment with TNFα. E2 protected against the TNFα-induced decrease in TER in a dose-dependent manner, and this protective effect was reversed by co-treatment with ICI 182,780 (Figure 3A). The cell viability did not differ significantly in any of the experimental groups (Figure 3B). Changes in the levels of the JAM-A and ZO-1 proteins reflected the changes in the TER. E2 protected against the TNFα-induced decrease in ZO-1 levels, and this protective effect was reversed with the co-treatment with ICI 182,780. The levels of JAM-A showed a similar pattern to that of ZO-1, but no statistical significance was observed (Figure 3C and D). These results indicate that E2 can protect against the TNFα-induced impairment of the oral epithelial barrier function in a nuclear ER-dependent manner.
 | Figure 3Protective effect of E2 on the TNFα-induced damage in oral epithelial physical barrier. Tight-junctioned monolayers of human oral keratinocyte (HOK-16B) cells were pre-treated with either ICI 182,780 for 6 hours and/or E2 for 4 hours and then treated with 100 ng/mL TNFα for 24 hours. (A) The TER was measured at the end point and expressed as the percentage of baseline. (B) Cell viability was measured using the CCK-8 assay kit at 24 hours. (C) After fixation, cells were stained for JAM-A and ZO-1 and examined by confocal microscopy. (D) The fluorescence intensities of JAM-A and ZO-1 were analyzed.E2: estradiol, TER: transepithelial electrical resistance, TNFα: tumor necrosis factor alpha, CCK: cell counting kit, JAM: junctional adhesion molecule, ZO: zonula occludens, TJ: tight junction.
a)P<0.05 versus no treatment (1-way analysis of variance with the Tukey post hoc test).
|
E2 inhibits the TNFα-induced nuclear translocation of NF-κB
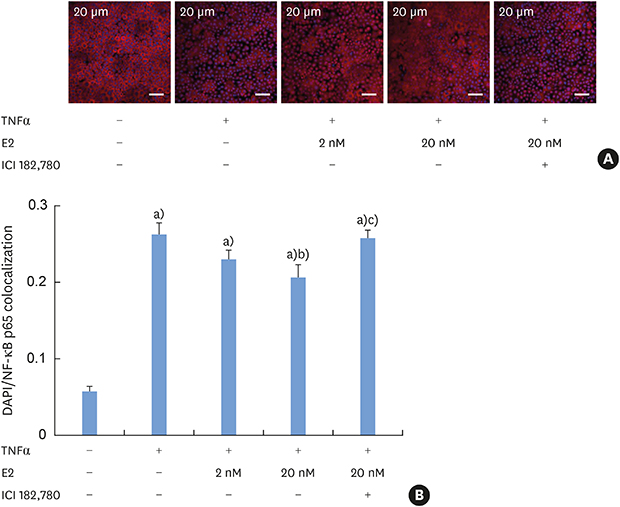
To understand the mechanism of the antagonism of TNFα by E2 in the regulation of the epithelial barrier function, the effect of E2 and ICI 182,780 on TNFα-induced NF-κB nuclear translocation was observed by confocal microscopy. Treatment with TNFα induced NF-κB nuclear translocation in 30 minutes. Pre-treatment with E2 reduced the level of TNFα-induced NF-κB nuclear translocation in a dose-dependent manner, and this effect was completely reversed by co-treatment with ICI 182,780 (Figure 4). These results indicate that E2 antagonizes TNFα-induced NF-κB nuclear translocation.
 | Figure 4Inhibition of the TNFα-induced nuclear translocation of NF-κB by E2 (A) Tight-junctioned monolayers of human oral keratinocyte (HOK-16B) cells were pre-treated with either ICI 182,780 for 6 hours and/or E2 for 4 hours and then treated with 100 ng/mL TNFα for 30 minutes. After fixation, cells were stained for NF-κB p65 and examined by confocal microscopy. (B) Co-localized signals of nucleus and NF-κB p65 were measured.E2: estradiol, TNFα: tumor necrosis factor alpha.
a)P<0.0001 versus no treatment; b)P<0.05 versus treatment with TNFα alone; c)P<0.01 versus treatment with TNFα and 20 nM E2 (1-way analysis of variance with the Tukey post hoc test).
|
E2 and corticosteroids act additively on barrier formation in oral epithelial cells
Corticosteroids, the drug of choice for diverse oral inflammatory mucosal diseases, have a well-known protective effect on epithelial barrier function [21]. We investigated whether E2 and corticosteroids could synergistically enhance the barrier function of the oral epithelium. Treatment with 250 nM Dexa alone increased the TER to a similar level as 20 nM E2, and co-treatment with Dexa and E2 had an additive effect on the TER (Figure 5A). The viability was not affected in any experimental conditions (Figure 5B).
 | Figure 5Additive effect of E2 and Dexa on the barrier formation in oral epithelial cell monolayers (A) Human oral keratinocyte (HOK-16B) cells seeded on the membrane of a transwell 2-chamber tissue culture system were treated with 20 nM E2, 250 nM Dexa, or E2+Dexa during barrier formation. The TER was measured at the indicated time points. (B) The proliferation of HOK-16B cells at 24 h was measured using the CCK-8 assay kit.E2: estradiol, Dexa: dexamethasone, TER: transepithelial electrical resistance, CCK: cell counting kit.
a)P<0.05 and b)P<0.0001 versus vehicle control (1-way analysis of variance with the Tukey post hoc test).
|
DISCUSSION
In this study, we report that E2 reinforced the physical barrier of oral epithelial cell monolayers by increasing the levels of TJ proteins in a nuclear ER-dependent manner. E2 increased the TER of HOK-16B cell monolayers in a dose-dependent manner during barrier formation, which was accompanied by increases in the levels of the JAM-A and ZO-1 proteins but no increase in cell proliferation. In addition, E2 protected HOK-16B cell monolayers against the TNFα-induced decreases in the TER and in the levels of ZO-1. The complete reversal of the effect of E2 by ICI 182,780 indicates that the effect of E2 on the barrier function of HOK-16B cell monolayers was mediated through nuclear ER, ERα, or ERβ. Because oral epithelial cells express only ERβ [18], ERβ must have mediated the function of E2 in HOK-16B cells. These results agree with earlier studies performed in other epithelia. E2 benzoate was shown to decrease the colonic paracellular permeability of ovariectomized rats by up-regulating occludin and JAM-A through ERβ signaling [16]. Similarly, E2 attenuated the acid-induced decrease in TER and the appearance of dilatation of the intercellular space in esophageal epithelia by up-regulating occlusion [17]. Therefore, estrogens seem to have a similar barrier-enforcing effect on various epithelia, including the oral epithelium, through ERβ.
In studies using intestinal Caco-2 or HT-29/B6 cells and pharmacological inhibitors, it has been shown that the TNFα-induced modulation of intestinal epithelial TJ proteins and barrier function depends on the activation of NF-κB [222324]. E2 inhibited the TNFα-induced nuclear translocation of NF-κB in HOK-16B cells, and this effect was completely reversed by ICI 182,780. This suggests that ERβ-mediated inhibition of NF-κB may play an important role in the protective effect of E2 on TNFα-induced barrier dysfunction in oral epithelium. In addition to classical ER-dependent transcriptional regulation through estrogen response elements, estrogens exert their function through diverse mechanisms [14]. Inhibition of phosphorylation and degradation of IκBα by E2 has been shown in rat brains and HeLa cells overexpressing ER, respectively [2526]. Alternatively, E2 inhibited TNFα-induced processing of NF-κB p105, the NF-κB precursor that blocked the translocation of NF-κB p50 and p65 in MCF-7 breast cancer cells [27]. In addition, direct interactions between ERα and NF-κB in the nucleus have been shown in osteoblastic U2-OS cells [28]. In the nucleus, ER proteins may directly inhibit NF-κB DNA binding, as shown in vitro [29] or indirectly by interfering with the interaction between NF-κB and transcriptional coactivators [30]. In our study, 20 nM E2 inhibited the TNFα-induced nuclear translocation of NF-κB only by about 30%, but the decrease in TER was by about 78%. Therefore, ER seems to interfere with the nuclear translocation of NF-κB but also seems to antagonize the function of NF-κB within the nucleus of oral epithelial cells.
Dexa gargle is currently the treatment of choice for several inflammatory oral mucosal diseases, such as oral lichen planus and recurrent aphthous stomatitis. E2 and Dexa showed an additive effect on oral epithelial barrier formation. Furthermore, E2 has been reported to exhibit anti-inflammatory activity in diverse tissues [313233]. Because E2 is cell-permeable and ERβ is expressed in all cell layers of the stratified squamous oral epithelium [18], E2 can be topically applied to minimize potential side effects. The mixture of E2 and Dexa may be more effective in treating inflammatory oral mucosal diseases than Dexa alone. In that case, the dose of Dexa can be lowered and E2 may to reduce the side effects of Dexa.
One of the limitations of the current study is that the detailed molecular mechanisms of TJ regulation in oral epithelial cells were not determined. Miyagawa et al. [34] reported that the TNFα-induced decrease in the TER in human gingival epithelial cells recovered in response to p38 MAPK and ERK inhibitors though upregulation of E-cadherin. Although the PKC, MAPK, and NF-κB signaling pathways are known to be involved in the expression of TJ proteins and the regulation of the barrier function in various organs, the complete signaling pathways that regulate TJs remain unclear [13]. One of the common side effects of the prolonged use of Dexa gargle is oral candidiasis, which may be attributed to the down-regulation of cathelicidin, a candidacidal antimicrobial peptide, by Dexa [3536]. It has been reported that an ERβ ligand, genistein, stimulates cathelicidin expression in human keratinocytes through the production of S1P and the subsequent activation of NF-κB and C/EBPα [37]. Delineation of signaling pathways that regulate TJs, AJs, and antimicrobial peptides in future studies may enable the specific regulation of the epithelial barrier while preserving protective innate immune responses.
Another limitation of the current study is that the effect of E2 on barrier function was examined in cell monolayers, although oral epithelial cells form a stratified squamous epithelium in vivo. Most current knowledge on the regulation of epithelial barrier function by TJs and AJs is derived from simple epithelium, such as the intestinal epithelium or cell monolayers. Even the 3-dimensional distribution of TJs and AJs in the stratified squamous epithelia is not clearly known. How the regulation of the epithelial barrier in the stratified epithelia is different from that in cell monolayers warrants further study.
In conclusion, E2 reinforces the physical barrier of oral epithelial cells through the nuclear ER-dependent upregulation of TJ proteins. The protective effect of E2 on the TNFα-induced impairment of the epithelial barrier and the additive effect of E2 with Dexa suggest its potential use to treat oral inflammatory diseases involving epithelial barrier dysfunction.
 XML Download
XML Download