

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Medications are generally considered as the first line of treatment in dystonias. Unfortunately, anticholinergics, benzodiazepines, muscle relaxants, anti-dopaminergic drugs, and dopaminergic drugs are not very effective in most dystonias. Therefore, chemo-denervation by botulinum toxin injection or surgical treatments such as myotomy, rhizotomy, and deep brain stimulation are often applied for treatment of dystonia.12
However, there is a special group of disorders that show good responses to dopaminergic drugs. Dopa-responsive dystonia (DRD) is a classic example.34 Its classic features are summarized as childhood or adolescent onset dystonia associated with marked diurnal fluctuation and improvement by sleep or rest, and a dramatic and sustained response to low doses of dopaminergic drug.4 Typical picture of DRD was first described by Segawa et al.5 with the name of “hereditary progressive basal ganglia disease with marked diurnal fluctuation” and later "hereditary progressive dystonia with marked diurnal fluctuation.”67
In 1986, Nygaard and Duvoisin8 collected similar cases and reported that most cases had also parkinsonism as well as dystonia. At that time, idiopathic torsion dystonia was the most common young-onset dystonia and was rather refractory to many medications tried including levodopa. They noted that diurnal fluctuation in DRD is not always present and does not reliably distinguish DRD from idiopathic torsional dystonia. They also noted that DRD can present with a dystonic gait disorder, diplegic cerebral palsy, sporadic spastic paraplegia, ataxic syndromes, and juvenile parkinsonism in children and adolescents. They emphasized that the response to levodopa is so dramatic and occurs so quickly that a therapeutic trial for diagnosis should be undertaken in all patients presenting with these syndromes. Since then, they coined the term the “dopa-responsive dystonia” to emphasize levodopa responsiveness.9 Even though there have been many reports of similar clinical presentations with the different names,1011121314151617 the term “dopa-responsive dystonia” introduced by Nygaard et al.9 seems to be most widely used nowadays.
However, many reports have shown atypical or incompatible features under the name of DRD such as infantile onset, psychomotor retardation, convulsion, hypotonia, drowsiness, hyperthermia, ptosis, cerebellar dysfunction, incomplete responsiveness to levodopa, etc.4181920 On the other hand, mutations in dopamine synthetic enzymes other than GTP cyclohydrolase I (GCH-1) resulted in similar clinical manifestations according to the severity of enzymatic defects.4 These clinical and genetic heterogeneities were enough to confuse clinicians in making the diagnosis.
With this background, we proposed the concepts of DRD and DRD-plus in 1998 and 2014 to embrace the clinical and genetic heterogeneities.421 DRD was defined as a syndrome of selective nigrostriatal dopamine deficiency caused by genetic defects in the dopamine synthetic pathway without nigral cell loss; and DRD-plus as a group of disorders caused by genetic defects in the dopamine synthetic pathway without nigral cell loss that have features of DRD ‘plus’ those features that are not seen in DRD. DRD-plus patients have more severe enzymatic defects which result not only in dopamine deficiency, but also in serotonin and/or norepinephrine deficiency. This pathophysiologic feature is also closely linked to the variable responses to dopaminergic medications as well as to the clinical manifestations. DRD patients generally show marked improvements with only small dose of dopaminergic drugs. In contrast, dopaminergic drugs are only partially effective in DRD-plus. Additional supplement of serotonergic and adrenergic medications may be necessary depending on the type and severity of enzymatic defects. The concepts of DRD and DRD-plus make differential diagnosis practical and easy, contribute to guiding therapeutic plan, and help expect responses to medications.4
However, there have been many reports of dystonia responsive to dopaminergic drugs, which do not fit into DRD or DRD-plus. For example, the transportopathies such as dopamine transporter (DAT) deficiency and vesicular monoamine transporter 2 (VMAT2) deficiency have similar characteristics to DRD-plus.2223 However, they are caused by the defects in dopamine reuptake, not in the dopamine synthesis. Some neurodegenerative disorders which involve nigrostriatal dopaminergic system such as juvenile Parkinson's disease (JPD), pallidopyramidal syndrome (PPS), and spinocerebellar ataxia type 3 (SCA3) also mimicked features of DRD or DRD-plus.2425262728293031 Moreover, there are reports of cases with DYT1 mutation, GLUT1 mutation, myoclonus-dystonia (MD), and ataxia telangiectasia (AT) under the heading of DRD because of their dopa-responsiveness, however, these disorders do not involve nigrostriatal dopaminergic system.32333435
Herein, we expand the concept of DRD and DRD-plus to include cases described above and formulate an etiological classification to help make a differential diagnosis and plan treatment.
CLASSIFICATION OF DYSTONIAS RESPONSIVE TO DOPAMINERGIC DRUGS
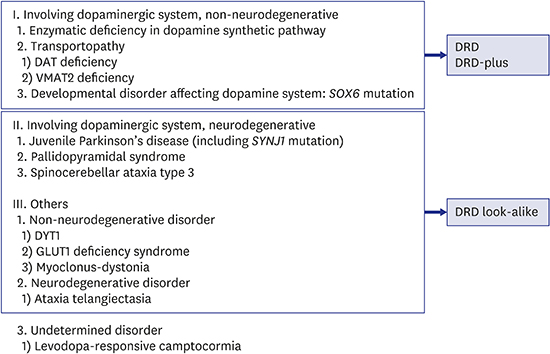
We categorized the disorders with dystonias which are responsive to dopaminergic drugs based on 2 criteria: 1) the presence or absence of involvement of dopaminergic system; and 2) the presence or absence of evidence for neurodegeneration (Table 1). If any dystonias are responsive to dopaminergic drugs, the involvement of dopaminergic system is suspect. Actually, the disorders with defects in dopaminergic system account for the majority with dystonias responsive to dopaminergic drugs. However, there are reports of disorders without evidences of involvement of dopaminergic system but still responsive to dopaminergic medications, even though they are rare and difficult to understand.32333435
Table 1
Classification of dystonias responsive to dopaminergic drugs

![]()
Whether the disorders are caused by neurodegenerative change is also important in diagnostic approach. The neurodegenerative disorders are progressive in nature even though the initial presentation may be similar to that of non-neurodegenerative disorders. The cases of dopa-responsive camptocormia are classified as the undetermined group because there are no clues to judge by the above 2 criteria.3637
NON-NEURODEGERATIVE DISORDER WITH INVOLVEMENT OF DOPAMINERGIC SYSTEM
Enzymatic defects in dopamine synthesis
It was amply discussed in our previous publications on this topic, and will be presented only in brief.42138 The enzymatic defects in dopamine biosynthetic pathway can be divided into 3 sub-parts according to the step: 1) the enzymatic defects in synthesizing tetrahydrobiopterin (BH4) (GTP cyclohydrolase I [GCH-1], 6-pyruvoyltetrahydropterin synthase [PTPS], sepiapterin reductase [SR], dihydropteridine reductase [DHPR]), 2) the enzymatic defect in synthesizing levodopa (tyrosine hydroxylase [TH]), 3) the enzymatic defect in synthesizing dopamine and serotonin (aromatic L-amino acid decarboxylase [AADC]) (Fig. 1). Theoretically, BH4 deficiency may cause serotonin deficiency as well as dopamine deficiency. However, in cases with DRD phenotype, symptoms for serotonin deficiency appear infrequently because TH has the higher Km for BH4 in the Michaelis-Menten kinetics.39 When the degree of BH4 deficiency is more severe, serotoninergic system are also affected.
 | Fig. 1Characteristics of enzymatic deficiencies on dopamine synthetic pathway.GTP = guanosine triphosphate, GCH-1 = GTP cyclohydrolase 1, NH2P3 = dihydroneopterin triphosphate, NP = neopterin, PTPS = 6-pyruvoyltetrahydropterin synthase, 6-PPH4 = 6-pyruvoyl-tetrahydropterin, SR = sepiapterin reductase, BH4 = tetrahydrobiopterin, DHPR = dihydropteridine reductase, qBH2 = quinonoid dihydrobiopterin, Tyr = tyrosine, Trp = tryptophan, Phe = phenylalanine, TH = tyrosine hydroxylase, TPH = tryptophan hydroxylase, PAH = phenylalanine hydroxylase, L-dopa = levodopa, 5-HTP = 5-hydroxydryptophan, AADC = aromatic L-amino acid decarboxylase, NE = norepinephrine, HVA = homovanillic acid, 5-HIAA = 5-hydroxyindolacetic acid.
|
Among enzyme deficiencies in dopamine synthetic pathway, DRD phenotype was most often reported in autosomal dominant (AD) GCH-1, and rarely in autosomal recessive (AR) SR, and TH deficiencies.38 AD GCH-1 deficiency generally shows less severe deficiency than AR GCH-1 deficiency because only one allele is affected in AD GCH-1 mutation. The enzymatic defect in SR deficiency is also less severe because of the alternative pathway (Fig. 1).
GCH-1 mutation
AD GCH-1 deficiency have one mutated allele so that its enzymatic defect is milder than that of AR GCH-1 deficiency. It presents as childhood-onset dystonia. In general, there is no mental retardation, seizure, myoclonic attack, or autonomic dysfunction.438 The response to levodopa is dramatic and long-term complications such as motor fluctuation and dyskinesia are infrequent. Even though there were several cases with levodopa-induced dyskinesia (LID), it disappears with time40 or by adding amantadine.41 Most of AD GCH-1 deficient cases showed good response at 100–300 mg/day of levodopa.38 Some patients with AD GCH-1 mutation were reported to have residual symptoms even with levodopa treatment.42
AR GCH-1 deficiency have 2 mutated copies, and are more severe in phenotypes than those of AD GCH-1 deficiency group. Earlier (neonatal or infant) onset, more aggressive motor symptoms (hypotonia, difficulties in swallowing and locomotive movements, etc.), and developmental delay have been reported.1843 Many patients with AR GCH-1 mutation tend to respond only partially to levodopa.38 However, some cases with homozygous mutations presented as classic DRD with excellent response to levodopa and mild residual symptoms but the earlier age of onset.4344 It was suggested that the severity of functional impairment, not the number of mutation, determines the phenotype.
PTPS mutation
AR PTPS mutated cases were reported with early onset and very severe phenotypes.45 The recent systematic review noted that motor outcomes of the cases with PTPS mutation were poorer than GCH-1, SR, and TH mutations.38 However, early diagnosis and intervention with replacement therapy led to much better prognosis.46 In addition, Leuzzi et al.45 reported that delayed treatment led to poor outcome with mental retardation, emphasizing the need to be aware of these disorders even though quite rare.
SR mutation
The motor outcome of SR deficiency is as excellent as that of AD GCH-1 deficiency38 because the alternative pathway can alleviate the severity of BH4 deficiency (Fig. 1). Thus the many of SR deficient cases belong to DRD phenotype. However several cases presented as DRD-plus phenotype. Bonafé et al.47 reported 2 cases who showed dystonia, spasticity, oculogyric crisis, tremor, ataxia, and psychomotor/growth retardation with marked diurnal fluctuation. The second patient had permanent structural change in nervous system such as microcephaly. The homozygous (c.1303–1304TC→CT) and compound heterozygous (c.1397A→G, 1397–1401delAGAAC) mutations were found respectively.
DHPR mutation
DHPR is a recycling enzyme of BH4 (Fig. 1). Most patients with DHPR mutation had near complete deficiencies of enzyme activity and showed DRD-plus phenotypes. Ponzone et al.48 reported the characteristics: 4 to 6 months of median onset age; low birth weight; microcephaly; hypotonia; drowsiness; swallowing difficulties; abnormal movements; oculogyric crises; convulsions; and recurrent episodes of hyperthermia. The clinical outcomes of DHPR deficient cases were relatively poor like those of PTPS deficient cases.38
TH mutation
There is no serotonin deficiency in patients with TH mutation (Fig. 1), but the symptoms caused by NE deficiency such as ptosis, miosis, and postural hypotension can occur depending on its severity.4 Its clinical spectrum includes typical DRD,49 dopa-responsive infantile parkinsonism,50 and progressive infantile encephalopathy.5152 Progressive infantile encephalopathy and dopa-responsive infantile parkinsonism clinically belong to DRD-plus category. Mental retardation, ocular dysfunction, parkinsonism, myoclonic jerk, and ptosis were reported. The responsiveness to levodopa or phenylephrine was also restricted.438
AADC mutation
AADC is a critical enzyme at the last step in producing dopamine and serotonin (Fig. 1). The AADC mutated cases generally showed DRD-plus presentation. Brun et al.53 summarized common symptoms and signs of 78 AADC deficient cases. The onset of age was infancy in 38 patients (48.7%) of them. Hypotonia were most common symptoms (74 of 78 patients, 95%) rather than dystonia (41 patients, 53%) throughout all age group. Oculogyric crisis, developmental retardation, ptosis, etc. were also frequently reported.
Levodopa and 5-hydroxydryptophan, precursors of dopamine and serotonin, did not improve the clinical symptoms in this disorder. Dopamine agonists and monoamine oxidase inhibitors could be applied but with insufficient effects.38 The results for treatments seem to be most unfavorable among enzymatic deficiencies in dopamine synthetic pathway. Even with medical treatments, most patients inevitably face non-ambulatory, non-verbal, and tube-fed outcome.54
Transportopathy
The transporters are vital in recycling monoamines. Thus, the defects of transporters lead to monoamine deficiency. Two transportopathies have been reported and the clinical characteristics were nearly compatible with DRD-plus.2223
DAT deficiency
DAT recycles released dopamine into presynaptic nerve terminals, and its defect leads to dopamine depletion in dopaminergic nerve terminals. Kurian et al.2255 reported a series of patients who had infantile onset parkinsonism-dystonia. During childhood, patients developed severe parkinsonism-dystonia accompanied with an eye movement disorder, pyramidal tract features, and motor developmental delay. Parkinsonism, dystonia, developmental delay developed before 3 years of age. All patients showed an increased ratio of homovanillic acid (HVA) to 5-hydroxyindoleacetic acid (5-HIAA) in CSF analysis. Urine HVA level was also slightly increased in 6 of 7 patients evaluated. DAT scan showed virtual absence of DAT in the striatum. Levodopa in small dose was effective only in 2 patients among 9, but dyskinesia developed in higher doses, which improved with dopamine agonists. The others did not get any beneficial effects from levodopa. Dopamine agonists provided partial improvement in a few patients. Thus, main clinical features were similar between DAT deficiency and severe dopamine synthetic enzyme defects. But ocular flutter and saccadic initiation defect were characteristics of DAT deficiency, whereas microcephaly, seizure, and oculogyric crisis were more common in dopamine synthetic enzyme disorders.22
VMAT2 deficiency
VMAT2 transports monoamines from cytosol into synaptic vesicle, and its defect results in depletion of monoamines in the nerve terminals.56 Rilstone et al.23 reported 8 children with SLC18A2 mutation, which encodes VMAT2, from a consanguineous Saudi Arabian family inherited in an AR fashion. The index patient had hypotonia at 4 months of age. When she visited clinic at 16 years of age, she had developmental delay, oculogyric crisis, excessive diaphoresis, profuse nasal secretion, hypotonia, dysarthria, and ataxia. There was no diurnal fluctuation. At neurological examination, she showed ptosis, limited upward gaze, parkinsonism, hypotonia, and dystonia. The treatment with levodopa was not helpful, but dopamine agonists improved the symptoms with minimal side effects. Clinical symptoms and responsiveness to treatments of VMAT2 deficiency are very similar to those of AADC deficiency because both disorders significantly affect the generation of serotonin as well as of dopamine. However, lack of improvement with the Vitamin B6 (a cofactor of AADC) and absence of diurnal fluctuation of VMAT2 deficiency are different from AADC deficiency.23
Developmental disorder affecting dopamine system: SOX6 mutation
SOX6 gene has a critical role in regulating the differentiation of nigral dopaminergic neurons, cortical interneurons, and oligodendrocytes.575859 Its mutation results in dysfunctions of diffuse cortical and basal ganglia areas. There is no evidence of neurodegeneration.
Ebrahimi-Fakhari et al.60 reported a 15-year-old male presented with acute-onset and rapid progressive generalized dystonia, athetoid movement, tremor, and dysarthria. The patient had development delay and attention deficit-hyperactivity disorder. There was no diurnal fluctuation. Physical exam was significant for dysmorphism, splenomegaly, sternal deformity, and slow horizontal/vertical saccades as well as dystonia, athetoid movement, and resting/action tremor. The work-up showed de novo deletion including SOX6 gene. The effect of 300 mg/day of levodopa was rapid and remarkable, which was sustained until last follow-up of age 20 years. Although DAT imaging was not done in SOX6 mutation, the experimental study showed that SOX6 gene is linked to development and survival of dopaminergic neurons in the substantia nigra.57
NEURODEGENERATIVE DISORDER WITH INVOLVEMENT OF DOPAMINERGIC SYSTEM
Several neurodegenerative disorders affecting dopamine system can present with dystonias responsive to dopaminergic drugs. JPD (including SYNJ1 mutation),2425262728 PPS,2930 and SCA331 are good examples. They have frequent parkinsonism, relatively late onset, and progressive disease course with drug-related complications compared to non-neurodegenerative disorders in the dopaminergic system.
Juvenile Parkinson's disease
JPD is a well-known disorder which can present as dystonia in the leg with diurnal fluctuation and initial excellent response to dopaminergic drugs.24252627 Thus, JPD is an important differential diagnosis from DRD.4 However, progressive degenerative cell loss in JPD leads to long-term motor complications and requiring higher levodopa dose with time. DAT imaging can make an easy differential diagnosis.
SYNJ1 mutation could cause a severe form of JPD. It is associated with parkinsonism, dystonia, hypotonia, resting/action tremor, postural instability, seizure and cognitive impairment.2861 The age at onset is around 20 years and the symptoms are progressive. The presynaptic nigrostriatal dopaminergic deficits in DAT scan and levodopa responsiveness for parkinsonism were reported in several cases.626364
Rauschendorf et al.28 reported two siblings with generalized dystonia. They had the histories of seizure in the childhood. They showed dramatic response to a small dose of levodopa, and one of them developed dyskinesia with the increase of dose. DAT scan showed presynaptic nigrostriatal dopaminergic deficits in both cases. Although the gene studies for TH, GCH-1, and SR were performed with suspicion of DRD, those were negative. The final diagnosis was made as SYNJ1 mutations by next-generation sequencing.
Sometimes, a special consideration is necessary in co-existing cases of DRD and JPD as Hjermind et al. 65 reported. At age 28, the patient presented as DRD phenotype. Mutation in GCH-1 gene was found with decreased enzyme activity. At age 35, dyskinesia developed which is infrequent in classic GCH-1 mutation. DAT imaging showed decreased DAT binding which is compatible with PD. Therefore, this degenerative portion will determine his ultimate prognosis. However, further discussions are required to determine whether it was just co-incidence or GCH-1 mutation acted as a risk factor for PD, because it is recently reported that GCH-1 mutations are associated to an increased risk for PD.66
Pallidopyramidal syndrome
PPS includes several genetic disorders.67 Kara et al.68 redefined PPS which are presented as Davison's triad (parkinsonism, dystonia, and spasticity) with or without iron accumulation on MRI. For the treatment of dystonia, symptomatic approach with medications is main strategy in PPS.69 Although anticholinergics, benzodiazepines, and muscle relaxants have been used frequently, some cases showed good response to levodopa. Sina et al.29 reported two families with PLA2G6 point mutation presenting as dystonia-parkinsonism responding to levodopa. Paisán-Ruiz et al.30 reported that not only PLA2G6 but also ATP13A2, FBX07, and SPG11 mutations presented with some dystonia with partial levodopa response. The age of onset mainly ranged from childhood to adolescence. In addition, there were developmental delay, neuro-psychiatric symptoms, pyramidal signs and partial responsiveness to levodopa like in DRD-plus phenotype.
Spinocerebellar ataxia type 3
Although SCA3 is a genetic ataxic syndrome, it can also have parkinsonism, dystonia, spasticity, peripheral neuropathy, and neuro-psychiatric symptoms.707172 Wilder-Smith et al.31 reported a case of adult onset leg dystonia with marked diurnal fluctuation. The age of onset was 37 years and her symptoms gradually progressed for 3 years. Brain MRI and CSF profile did not reveal any remarkable findings. A small dose of levodopa (100 mg daily) showed marked improvement, lasting for 3–5 hours. The initial assessment was DRD. However, there was a family history over 5 generations with ataxia, peripheral neuropathy and SCA3 was confirmed later.
DISORDER WITHOUT INVOLVEMENT OF DOPAMINERGIC SYSTEM
As noted above, it is poorly understood why the disorders in this category show dopa-responsiveness. There have been only a few case reports.32333473
Non-neurodegenerative disorder
DYT1 mutation
Dystonia associated with DYT1 mutation generally begins at around age 13.74 About 60% of DYT1-mutated patients tend to progress into generalized form. Dystonia with DYT1 mutation typically does not respond to levodopa. However, there was a report with dystonia in upper extremities responding to levodopa and trihexyphenidyl.33 Postural and action dystonia of both upper extremities began in right hand at age 7 and in left upper extremity at age 9. Levodopa and trihexyphenidyl improved postural dystonia in the right hand. With further dose escalation of levodopa, both types of dystonia in the right and left arms were well ameliorated. At age 11, GAG deletion in the DYT1 gene was confirmed
GLUT1 mutation
Glucose transporter 1 (GLUT1) deficiency results in impaired glucose transport into the brain.75 The classic cases are characterized by seizures, developmental delay, microcephaly, hypotonia, spasticity, ataxia, and dystonia. However, in some, main manifestation of GLUT1 mutations presents with paroxysmal exercise-induced dyskinesia.76 Ketogenic diet is a key treatment, and several medications such as valproate and acetazolamide might be helpful in symptom relief.
Baschieri et al.32 reported a case of paroxysmal exercise-induced dystonia due to GLUT1 mutation, which was responsive to levodopa. The patient (47 year-old male) visited the clinic with paroxysmal dystonic attacks since childhood. The symptoms lasted for 20 to 165 minutes and occurred daily on average. CSF to blood ratio of glucose was consistent with GLUT1 deficiency, and the mutation (p.R333Q) in the SLC2A1 gene was confirmed. Patient refused ketogenic diet. Trials with oxazepam, diazepam, carbamazepine, clonazepam, valproate, and acetazolamide were completely unhelpful. When levodopa reached a dose of 800 mg daily, there was a reduction of both frequency and intensity of the attacks. But, with reduction of the levodopa to 200 mg daily, his condition was much worsened.
Myoclonus-dystonia
Myoclonus-dystonia (MD) syndrome is a disorder characterized by myoclonus and dystonia.77 The symptoms can often improve with alcohol ingestion. Although many trials of medications were unsatisfactory, there were some reports showing beneficial effects of anticholinergics, benzodiazepines, levetiracetam, piracetam, and zonisamide.
Two cases of myoclonus-dystonia with SGCE mutation responsive to levodopa were also reported.34 The first case developed myoclonus and dystonia on his trunk and extremities since age 12. Although he had mild developmental delay as well as macrocephaly, low set ears, and low birth weight/height, he caught up quickly with his psychomotor milestones. The information about alcohol responsiveness was not known. Valproate improved symptoms, but induced hyper-ammonemia. Clonazepam and trihexyphenidyl slightly improved symptoms. Levodopa (800 mg daily) was added to clonazepam and trihexyphenidyl with significant improvement. The second case showed irregular distal tremor in both hands, which spread into arms and legs. There were also writer's cramp, mild head tilt, and action myoclonus which were improved by levodopa. SCGE deletions were confirmed in these 2 cases. The mechanism of levodopa responsiveness is unclear, although there was a report that epsilon-sarcoglycan is expressed in midbrain monoaminergic neurons.78
Neurodegenerative disorder
Ataxia telangiectasia
Schneider et al.35 reported familial dopa-responsive cervical dystonia. There were 4 patients with torticollis as initial manifestation. Among them, 3 patients were included in a single family. The age of onset ranged from 9 to 15 years. All of them showed dramatic responses to levodopa. Most probable diagnosis was classic DRD. The genetic tests for GCH-1, TH, and SPR were negative. Several years later, they reported ATM gene as the causative gene in this family by whole exome sequencing.79 But of note is that levodopa was not effective in 3 patients with dystonic AT in another report.73 Therefore, it is not quite clear whether dopa-responsiveness in the former cases by Schneider et al.3579 is related to AT.
UNDETERMINED DISORDER
Dopa-responsive camptocormia
Van Gerpen36 described a case of dopa-responsive camptocormia with diurnal fluctuation. The initial complaint was tightness and twist in his paraspinal muscles at age 48. On neurologic examination at age 49, he showed camptocormia without parkinsonism, ataxia, and pyramidal tract signs. Walking backward amazingly improved the symptom. Camptocormia dramatically responded to levodopa (600 mg daily) for at least 6 years without motor complications. Genetic screening for classic DRD was not performed.
The second report of dopa-responsive camptocormia was recently described.37 Gradual onset of truncal flexion on standing begun since his age of 50. The symptoms worsened in the evening compared to in the morning. Levodopa (375 mg daily) was steadily helpful in improving dystonic camptocormia for 2 years. The DAT scan was normal. There were no mutations in GCH-1, TH, and THAP1 gene. As etiologies have not been found in these reports of dopa-responsive camptocormia so far, they are grouped into undertermined disorder. However they could be moved to one of the above categories in the future.
DISCUSSION
DRD, DRD-plus, and DRD look-alike
We reviewed the disorders with dystonias responsive to dopaminergic drugs. All of those showed DRD or DRD-plus ‘phenotype.’ However, many disorders do not meet our previous criteria of DRD or DRD-plus.42138 Therefore, we would like to propose DRD as a group of non-neurodegenerative disorders by genetic defects involving nigrostriatal dopaminergic system with cardinal manifestations (namely, DRD phenotype) (Table 2). DRD-plus is also re-defined as a group of non-neurodegenerative disorders by genetic defects involving nigrostriatal dopaminergic system with dopa-responsiveness ‘plus’ additional features (namely, DRD-plus phenotype) that are not seen in DRD. Those additional features include infantile onset, developmental delay, psychomotor retardation, seizure, hypotonia, drowsiness, recurrent hyperthermia, ptosis, cerebellar dysfunction, poor responsiveness to levodopa or other dopaminergic drugs, etc. This new definition will include transportopathies and developmental defect such as SOX6. In summary, DRD and DRD-plus share commonalities in etiologies in that they are not neurodegenerative and involve nigrostriatal dopaminergic system, and the clinical severity of each disorder varies according to the degree of genetic defect.
Table 2
New definitions of DRD, DRD-plus, and DRD look-alike

| Previous name | Previous definition4,21 | New definition |
|---|---|---|
| DRD | A syndrome of selective nigrostriatal dopamine deficiency caused by genetic defects in the dopamine synthetica pathway without nigral cell lossa | A group of non-neurodegenerative disorders by genetic defects involving nigrostriatal dopaminergic system with cardinal manifestationsb (namely, DRD phenotype) |
| DRD-plus | A group of disorders caused by genetic defects in the dopamine synthetica pathway without nigral cell lossa that have features of DRD ‘plus’ those features that are not seen in DRD | A group of non-neurodegenerative disorders by genetic defects involving nigrostriatal dopaminergic system with dopa-responsiveness plus additional featuresc (namely, DRD-plus phenotype) that are not seen in DRD |
| DRD look-alike | - | A group of 1) neurodegenerative or non-neurodegenerative disorders without involving the nigrostriatal dopaminergic system or 2) neurodegenerative disorders with involving nigrostriatal dopaminergic system, that could present with dystonia responsive to dopaminergic drugs |
DRD = dopa-responsive dystonia.
aWhich are deleted in the new proposal to include additional disorders; bThose include dystonia and/or parkinsonism, and dramatic response to levodopa without long-term motor complications; cThose include infantile onset, developmental delay, psychomotor retardation, seizure, hypotonia, drowsiness, recurrent hyperthermia, ptosis, cerebellar dysfunction, poor responsiveness to levodopa or other dopaminergic drugs.
![]()
Then, we would like to propose “DRD look-alike” as a group of 1) neurodegenerative or non-neurodegenerative disorders without involving the nigrostriatal dopaminergic system or 2) neurodegenerative disorders with involving nigrostriatal dopaminergic system, that could present with dystonia responsive to dopaminergic drugs (Table 2). The disorders listed in DRD look-alike could also present with DRD or DRD-plus phenotype. Levodopa-responsive camptocormia will be left as undetermined as of now. These terminologies will help to make an etiological thinking and to guide the laboratory investigation (Fig. 2).

Diagnostic approach to DRD, DRD-plus, and DRD look-alike
The diagnostic approach should start from the clinical characteristics. According to whether the patient present as DRD or DRD-plus phenotype, the probable etiologies could be determined (Fig. 3 and Table 3). One important consideration is the frequencies of diseases. Most of diseases showing dystonia with dopa-responsiveness belong to disorders that involve nigrostriatal dopaminergic system. The rest are just several case reports with small number of subjects (Fig. 3 and Table 3).
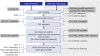 | Fig. 3Diagnostic flow for DRD and DRD-plus phenotypes.The disorders in the gray boxes are relatively common etiologies in each category. The DAT abnormality in the cases of SOX6 mutation has not been proven yet, but is very likely. Based on the experimental research, SOX6 gene is linked to development and survival of dopaminergic neurons in the substantia nigra.
DRD = dopa-responsive dystonia, [D] = DRD, [D+] = DRD-plus.
|
Table 3
Clinical and laboratory features of dystonias that are responsive to dopaminergic drugs

L-dopa = levodopa, DAT = dopamine transporter, DRD = dopa-responsive dystonia, Child = childhood, Adole = adolescence, NL = normal, Pro-NL = probably normal, AbNL = abnormal, VMAT2 = vesicular monoamine transporter 2, Pro-AbNL = probably abnormal, GLUT1 = glucose transporter 1.
aPro-NL and Pro-AbNL are presumptive descriptions based on patho-mechanism; bDystonia with dopa-responsiveness is only an infrequent manifestation in that disease.
![]()
If the patient presents as DRD phenotype, enzymatic deficiencies (due to GCH-1, TH, and SR mutations) in dopamine synthetic pathway and JPD should be initially considered (Fig. 3). DRD phenotype in SCA3, DYT1, GLUT1 deficiency, or AT is rare.313233 There were also 2 cases of dopa-responsive camptocormia presenting with DRD phenotype.3637
If the patient has DRD-plus phenotype, all enzymatic deficiencies in dopamine synthetic pathway, PPS, and JPD by SYNJ1 mutation can be considered in the differential diagnosis. Although transportopathies, SOX6 mutation, and MD can also present with DRD-plus phenotype,22233460 these are also infrequent. The age of onset and DAT scan would be very helpful in differential diagnosis (Fig. 3 and Table 3). Then, biochemical analysis and gene study will make a confirmative diagnosis.
DRD and Segawa disease
Name should reflect the characteristics of the disease. For example, clinical symptoms (e.g., myasthenia gravis), localizations (e.g., corticobasal syndrome), pathologic findings (e.g., dementia with Lewy body), and genetic backgrounds (e.g., DYT1 dystonia) contributes to making their names in neurological disorders. Eponyms tend to be changed into the new names which can cover the features of the diseases. For example, progressive supranuclear palsy is widely used nowadays instead of Steele-Richardson-Olszewski syndrome.
However, there are some exceptions. Parkinson80 reported PD as ‘shaking palsy.’ However, Charcot recognized that there are many PD cases without ‘shaking’ or ‘palsy,’ and used the name “Parkinson's disease,” which now includes not only motor but also non-motor features.
In his first report, Segawa et al.5 used the name ‘hereditary progressive dystonia with marked diurnal fluctuation’ which contains the important features of the disease. In 1994, Nygaard et al.9 used the term of “dopa-responsive dystonia” which also reflects the key features of dopa-responsiveness. However, as described in this review, many disorders were reported under the title of DRD when dopa-responsiveness was only partial and really did not resemble the classic DRD, thus confusing the picture of this unique disorder. Therefore, we would like to propose the new names: “Segawa disease” instead of “DRD”; “Segawa-plus” instead of “DRD-plus”; and “Segawa look-alike” instead of “DRD look-alike.” This nomenclature is clearer in giving accurate picture of the classic case of “DRD” and helps in logical thinking of etiology and diagnostic approach.
In conclusion, the updated definitions of DRD, DRD-plus, and DRD look-alike are more clinically oriented. At the same time, we believe that they can provide benefits in guiding the final diagnosis because the most cases of dystonia with dopa-responsiveness had genetic mutations (Table 3). Further studies with detailed clinical descriptions and genetic confirmations should be performed to clarify the boundaries between DRD, DRD-plus, and DRD look-alike and to understand the physiology of levodopa effectiveness to them.
 XML Download
XML Download