

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Wilson disease (WD) is an autosomal recessive disorder of Cu transport that has been described in every region of the world. It is the most common inherited liver disease, with a prevalence of 1:37,000 in the pediatric population of Korea.1 Mutations in the ATPase, Cu++ transporting, beta polypeptide (ATP7B) gene cause failure of Cu excretion into the bile and the defective incorporation of Cu into ceruloplasmin. Biochemical diagnosis is based on raised urinary Cu levels, low plasma ceruloplasmin levels, and elevated Cu concentrations in a liver biopsy.2 However, false positive rate of serum ceruloplasmin (< 20 mg/dL) was reported as 15% in Korean children.3 Some patients may be biochemically indistinguishable from healthy carriers4 and WD can sometimes be difficult to diagnose.
In 1993, the WD gene was cloned and shown to encode a P-type Cu transporting ATPase.5 Mutation analysis by direct DNA sequencing has been performed in many WD patients and genetic diagnosis is available in the clinical settings. In 1999, we reported that the p.R778L mutation is the most common mutation in Korean children with WD,6 and our investigations revealed high allele frequencies > 85% using polymerase chain reaction (PCR)-based direct DNA full sequencing.7 However, direct DNA sequencing cannot detect two mutations in every patient, which indicates a definite diagnosis. In previous studies, a 100% detection rate was never reached in patients with WD.8 So, the Ferenci score9 for the diagnosis of WD (scores ≥ 4) composed of clinical, biochemical, and genetic findings was introduced. This means that it is difficult to diagnose WD with only one definitive test.
Multiplex ligation-dependent probe amplification (MLPA) analysis is reportedly useful in increasing the diagnostic yield in other genetic disorders with large deletions or insertions that cannot be detected by conventional methods.1011 The MLPA analysis is based on the amplification of specifically hybridized probes up to 60 probes, each of which detecting a specific DNA sequence of approximately 60 nucleotides in length (http://www.mlpa.com). Each MLPA probe consists of two oligonucleotides that must hybridize to immediately adjacent target sequences in order to be ligated into a single probe.10 Each probe in an MLPA probemix has a typical amplicon length ranging between 130–480 nucleotides.12 According to the manufacturer's protocol, the SALSA 098 probemix (MRC-Holland, Amsterdam, the Netherlands) contains amplification products between 130 and 346 nucleotides. In contains 9 control fragments generating an amplification product smaller than 120 nucleotides, with four DNA Quantity fragments (Q-fragments) at 64-70-76-82 nucleotides, three DNA denaturation control fragments (D-fragments) at 88-92-96 nucleotides, one X-fragment at 100 nucleotides and one Y-fragment at 105 nucleotides (http://www.mlpa.com).
The aim of this study was to evaluate whether the detection rate of ATP7B mutations in patients with WD can be increased by using MLPA and to evaluate potential phenotype correlations.
METHODS
Study population
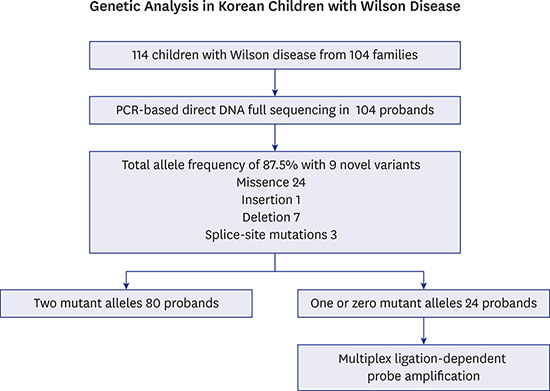
We enrolled 114 children from 104 families diagnosed with WD based on clinical and biochemical findings, and genetic tests by PCR-based direct DNA full sequencing at Seoul National University Children's Hospital. Clinical and biochemical findings of WD at diagnosis including the presence of Kayser-Fleischer ring, serum ceruloplasmin levels (< 0.2 g/L), urinary Cu concentrations (> 100 mcg/24 hr), and liver copper (> 250 mcg/d dry weight) contents were obtained from their medical records. The Ferenci score9 was calculated.
Direct DNA full sequencing of the ATP7B
PCR-based direct DNA full sequence analysis was performed for the genetic diagnosis of WD at clinical settings. Genomic DNA was extracted from 3–6 mL of EDTA anti-coagulated blood. The 5'-untranslated region and the whole 21 exon-coding region with flanking intronic sequences of the ATP7B gene were PCR-amplified using primers for each exon. The PCR products were subjected to direct double-stranded DNA sequencing of both the forward and reverse strands. The obtained sequences were compared with the reference sequence NG_008806.1 registered in the National Center for Biotechnology Information database (http://www.ncbi.nlm.nih.gov). Some of these patients have been reported previously, but only for total allele frequency.7
MLPA analysis
Of them, the patients with one or zero mutant allele were investigated using MLPA, using banked blood samples. MLPA was performed using SALSA MLPA® P098 kits (MRC-Holland). MLPA signal was interpreted using GeneMarker® software v1.70 (Softgenetics, State College, PA, USA). The ratios of test peaks to control peaks and control peaks to other control peaks in each sample were compared to the same ratios obtained for normal individuals. For normal sequences, a dosage quotient of 1.0 is expected. A deletion was scored if the mean dosage quotient of the test to control peaks was less than 0.65, and a duplication was scored if the mean dosage quotient was 1.40 or greater.
Statistical analysis
Analysis of variance and χ2 test were used to evaluate differences in clinical presentation and biochemical profiles between groups using Statistical Package for Social Science (version 20.0; SPSS Inc., Chicago, IL, USA).
Ethics statement
Informed consent was obtained from all individual's parents and/or children in the study. Banked blood samples which have been preserved under the approval of the Institutional Review Board of Seoul National University Hospital (approval No. 1102-099-357) were used. The present study protocol was reviewed and approved by the Institutional Review Board of Seoul National University Hospital (approval No. 1402-100-560).
RESULTS
Direct DNA full sequencing of the ATP7B
Mutations were identified in 182 of 208 independent alleles. The total allele frequency of ATP7B mutations in Korean children with WD was 87.5%. A total of 35 different mutations were found: 24 missense, 1 insertion, 7 deletion, and 3 splice-site mutations. The most frequent mutation, p.R778L accounted for 36.5% of all 208 studied WD alleles. The six other most common mutations were p.A874V, p.N1270S, p.L1083F, p.V872X, p.T1029I, and p.G1035V with allele frequencies of 9.62%, 7.21%, 6.25%, 4.32%, 3.37%, and 2.88%, respectively. Eleven variants were novel. Segregation tests were performed on the patients' families, and novel variants were not identified in 171 control samples (342 alleles). In silico analysis using PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2)13 predicted that p.H1069Y, p.Y1078C, and p.V1297I would probably be damaging with scores of 1.000, 1.000, and 0.871, respectively. p.I688S was predicted to be benign with a score of 0.157. Table 1 shows the ATP7B mutations detected using PCR-based direct DNA full sequencing. Ten more patients, who were siblings of the probands from nine families, were also investigated. Their genotypes and clinical presentations are listed in Table 2.
Table 1
ATP7B mutations in Korean children with Wilson disease detected using PCR-based direct DNA sequence analysis

![]()
Table 2
Sibling patients with Wilson disease and their characteristics

aMultiplex ligation-dependent probe amplification revealed no gross deletions or duplications in family 7.
![]()
A complete direct DNA full sequencing revealed a definite diagnosis of WD (2 mutant alleles) in 80 of 104 unrelated children with WD (76.9%). One mutant allele was detected in 22 children, and no mutations were detected in two children.
MLPA analysis and clinical characteristics of the children
MLPA analyses were performed on the 24 children with WD with one or zero mutant alleles. MLPA revealed no gross deletions (mean dosage quotient < 0.65) or duplications (> 1.40) in these 24 children. All of these 24 children had Ferenci scores ≥ 4 (range 4–9), which were highly diagnostic. Two children with no mutations showed serum ceruloplasmin levels of < 0.1 g/L, urinary Cu levels of 1,038, and 184 mcg/24 hr, respectively, and had Kayser-Fleischer rings. Among 24 children, serum ceruloplasmin levels were less than 0.1 g/L in 22 children, and 0.1–0.2 g/L in two children. Urinary Cu levels were > 100 mcg/24 hr in 23 children, and 85 mcg/24 hr in 1 child. Liver biopsies were performed in five children, with three showing more than 250 mcg/g Cu and two showing 50–250 mcg/g. These two children showed one mutant allele, serum ceruloplasmin levels less than 0.1 g/L, and one with urinary Cu levels of 179 and liver copper levels of 141 mcg/g, the other with urinary Cu levels of 85 mcg/24 hr, and liver copper contents of 167 mcg/g, respectively.
The clinical characteristics of the patients with one or zero mutant alleles and negative MLPA results did not differ from those of the patients with 2 mutant alleles, which indicate a definite genetic diagnosis of WD. The detection of 2 mutant alleles was not associated with hepatic manifestation, age of onset, presence of Kayser-Fleischer ring, serum ceruloplasmin levels, or urinary Cu concentrations (Table 3).
Table 3
Comparison of clinical and biochemical characteristics of Wilson disease between two groups based on genetic diagnosis

aMultiplex ligation-dependent probe amplification revealed no gross deletions or duplications in 24 children with one or zero mutation.
![]()
DISCUSSION
Since the ATP7B gene for WD was first reported in 1993, more than 800 different mutations have been described, including single base insertions and deletions, frame-shifts, missense, nonsense and splice-site mutations. The spectrum of WD mutations usually consists of a large number of rarely occurring mutations. Using PCR-based direct DNA sequencing of the ATP7B gene, we detected an allele frequency of 87.5% in our present study, and one or more mutations were identified in 98.1% (102/104) of the probands. To our knowledge, this is one of the highest detection rates yet reported.
The p.R778L missense mutation in exon 8 of the ATP7B gene was the most common molecular defect in our population, with a frequency of 36.5%. In concordance with previous studies from Japan (p.R778L, 27.7%)14 and China (37.7%),15 p.R778L is the most frequent mutation in East Asia. On the other hand, the p.H1069Q mutation, which is the most common mutation in Western populations, was not detected in our cohort. The second most common ATP7B mutation in Korean patients with WD, p.A874V, had a frequency of 9.62% in our cohort. This mutation was first described in Japan,16 but it is rare in Japan14 and China.15 These two mutations and five other common mutations account for 70.2% of all mutant alleles in WD patients. The remaining mutations are rare and observed in only a few patients.
However, not all patients with WD receive a definite genetic diagnosis. Thus, the Ferenci scoring system was developed at the International Meeting on WD to increase diagnostic accuracy.9 According to this system, patients with one or even zero mutant alleles could be diagnosed with WD if they have positive clinical and biochemical findings (Ferenci score of 4 or more). In 23% of our patients, we could not identify two mutant alleles, although they showed the characteristic clinical and biochemical features of WD. Whole exome sequencing revealed good concordance with direct DNA sequencing and would be timely effective,17 it also could not detect large deletion and duplications. It is possible that unknown molecular defects exist that are also be a cause of WD. There have been a case report of an Italian child born consanguineous with a deletion, c.51þ384_1708-953del spanning an 8798-bp region of the ATP7B gene from exon 2 to intron 4,18 and a case report of a Turkish child with a c.4021+87_4125-2del, is a 2144-bp deletion removing exon 20 and large parts of the flanking introns.19 MLPA is a highly sensitive method for detecting copy number variations. It can detect large deletions and duplications at the whole-exon level, which cannot found by direct sequencing. The combination of MLPA with DNA full sequencing increases the diagnostic accuracy in some genetic diseases such as Peutz-Jeghers syndrome10 or Alagille syndrome, which are known to have large gene defects. MLPA assays have been attempted in various pediatric diseases,202122 and few studies have assessed MLPA in WD patients. A deletion of exon 4 in 2 French patients with WD was reported, but no large gene rearrangements have been identified.23 Another study revealed no additional mutations.24 We found some deletions including novel deletions (frameshift mutations) by DNA full sequencing. However, our MLPA study revealed no additional gene defects in Korean children with WD, though 26 (12.5%) mutant alleles among 24 children have not been identified. All of those 24 children had Ferenci score > 4, which means diagnosis of WD. It suggests that large deletions or duplications in ATP7B might be extremely rare in patients with WD though few cases exist. The SALSA MLPA® 098 kit used in our study contains probes for 15 of the 21 exons of the ATP7B gene. It could not identify copy number variations for exons 9, 11, 12, 16, 19, and 21, and intragenic deletions which are in close proximity to neighbouring exons. Though the limitation exists, we can suggest that MLPA have little cost/benefit to increase the mutation detection rate in children with WD.
The clinical manifestations of WD are diverse, with wide variations in hepatic abnormalities and neurological disturbances. The assessment of whether the phenotypic diversity of WD is related to allelic heterogeneity is hampered by the large number of mutations identified in the ATP7B gene,25 and no definite association between genotype phenotype has been established. There have been many studies on the correlation of the p.H1069Q mutation with phenotype. Some studies have shown a clear correlation between the p.H1069Q homozygous genotype and neurologic presentation and a later onset of symptoms,262728 but this finding has not been reproduced by others.293031 The results of genotype-phenotype correlations might be different according to ethnicity, because the most frequent genotype is different in different racial groups. Okada et al.14 did not find any correlation between genotype and phenotype in Japanese patients. Wu et al.15 reported that homozygotes for the p.R778L mutation showed an earlier onset of symptoms with hepatic presentation in Chinese patients. Liu et al.32 showed a correlation between p.R778L and hepatic manifestations, but no correlation between p.R778L and an earlier onset of symptoms in Chinese patients with WD. We also analyzed genotype-phenotype correlations between the groups with p.R778L homozygous mutations, heterozygous mutations, and no p.R778L-related mutations in our Korean children with WD. The p.R778L mutation was not associated with age of onset, presence of Kayser-Fleisher ring, serum ceruloplasmin levels, or urinary Cu concentrations (data not shown). Most patients with WD usually have two different mutations rather than two identical mutations, so genotype-phenotype correlation studies are difficult to perform.
We analyzed the potential correlation between the number of detected mutant alleles and clinical manifestations; however, we could not identify any phenotypic correlation. The number of detected mutant alleles was not associated with clinical manifestation, age of onset, presence of Kayser-Fleischer ring, serum ceruloplasmin levels, and urinary Cu concentrations. Moreover, in our study, even within the same family, the modes of presentation were different. In a family with p.A874V/p.T1029I mutations, one child is presented with hepatic manifestation at 7 years of age, but the sibling presented neurologically at 19 years. In the other family with p.T1029I/- mutations, the proband presented neurologically, but his brother presented hepatically. MLPA revealed no additional deletions or duplications in these siblings. Phenotypic diversity in WD could not be explained by ATP7B mutations alone; thus, it is likely to due to additional factors.
Early diagnosis of WD is critical because treatment with chelating agents or oral Zn prevents liver cirrhosis and/or lifelong neurological disabilities. Since biochemical markers of impaired Cu metabolism can be misleading, and diagnosis may be difficult in the absence of typical symptoms and in asymptomatic siblings, genetic testing using PCR-based direct DNA full sequencing has played an important role in the diagnosis of WD. MLPA cannot give an additional benefit to increase the mutation detection rate in children who do not have a definite genetic diagnosis of WD by direct DNA full sequencing. This finding suggests that large gene defects in ATP7B a limited role in patients with WD. Further development of a novel technique is needed to improve the genetic diagnosis of WD.
 XML Download
XML Download