

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Immune-mediated glomerulonephritis (GN) includes the diseases that result from the deposition of immune complexes and/or complements in glomeruli.1 The deposition of complements without immune complexes in glomeruli is known as complement-mediated glomerulopathy, which was previously considered a subgroup of membranoproliferative GN. Complement-mediated GN, which includes C3 glomerulopathy, has been reclassified as dense deposit disease (DDD) (the presence of electron-dense osmiophilic material in the glomerular basement membrane) and complement-mediated GN (subendothelial and mesangial electron-dense deposits).2 In addition, Sethi group recently suggested C4 glomerulopathy, a new type of complement-mediated GN, with bright C4d staining and minimal or absent immunoglobulin and C3 staining on immunofluorescence (IF) microscopy.3 Sethi et al.3 have reported four cases of C4 glomerulopathy in the literature, one of C4 dense deposit disease (C4 DDD) and two of C4 GN.4 Herein, we present a case of C4d deposit-dominant GN, which occurred with non-infectious intermediate uveitis.
CASE DESCRIPTION
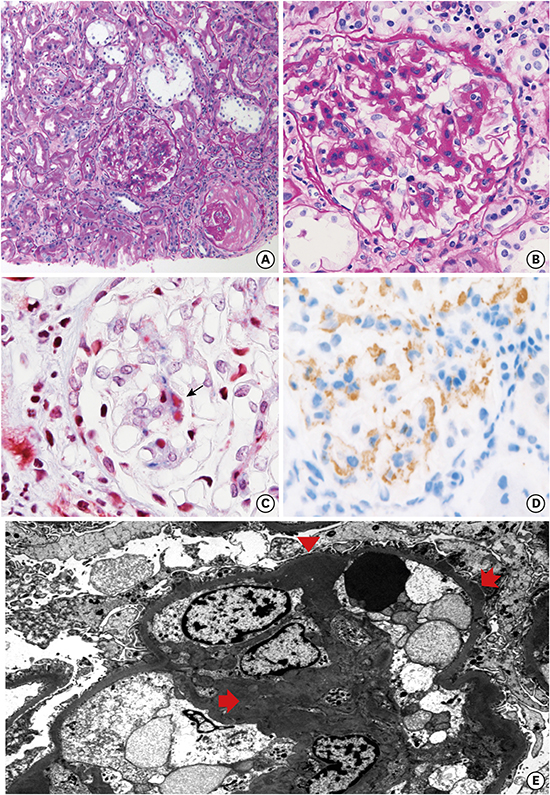
A 62-year-old man presented to the eye clinic on June 2016 with visual disturbance that had persisted for 2–3 months. On slit lamp examination, he had mild, bilateral, intraocular inflammation in the anterior chamber and vitreous. The inflammation was categorized as intermediate uveitis based on the clinical findings (Supplementary Fig. 1). To rule out systemic autoimmune disease, serologic and urinary evaluations were performed. His urinalysis showed hematuria and proteinuria. Therefore, the patient was referred to nephrology to evaluate the cause of the urinary abnormalities. Several laboratory tests for primary and secondary glomerulopathy were done. On a repeated urinalysis, he showed continuing hematuria (6–10 red blood cells per high power field) and proteinuria with protein excretion of 355 mg per gram of creatinine. His serum urea nitrogen (SUN) level was in the normal range, and his serum creatinine level was 1.22 mg/dL (corresponding to an estimated glomerular filtration rate of 63.1 mL/min/1.73 m2 as calculated by the Chronic Kidney Disease Epidemiology Collaboration). The C3 serum level was borderline low and the C4 serum level was normal. Serological evaluation for signs of infection, autoimmune disease, and monoclonal gammopathy produced negative results. On computed tomography, the size of both kidneys was shown to be in the normal range. Based on the laboratory results, the physicians were suspicious of primary glomerulopathy. So, a diagnostic kidney biopsy was performed. The kidney biopsy specimen contained 20 glomeruli, of which three were globally sclerosed (Fig. 1). Under light microscopy, five glomeruli exhibited proliferative histologic changes; three of them showed mild to moderate mesangial hypercellularity, and two of them showed fibrous crescents. The interstitium showed patch periglomerular fibrosis. Inflammatory cells, most of which were lymphocytes, had infiltrated into the fibrosis area. Eosinophils were not seen. Interstitial fibrosis and tubular atrophy were of moderate grade (approximately 50% for the total cortex). Under electron microscopy (EM), moderate electron-dense subendothelial, mesangial, and mild subepithelial deposits were identified. Congo red staining was negative. The IF results for the glomerular deposits were negative for anti-IgG, anti-IgA, anti-IgM, anti-kappa light chain, anti-lambda light chain, anti-albumin, anti-C3, anti-fibrinogen, and anti-C1q; however, the glomerular deposits were highlighted by anti-C4d immunohistochemistry (IHC) (rabbit polyclonal antibody; Cell Marque, Rocklin, CA, USA).
 | Fig. 1A case of GN with C4d deposits. (A) Light microscopy of glomerulus showing an increase in mesangial matrix with segmental mild hypercellularity (PAS, 100×). (B) A representative glomerulus showing mesangial widening (PAS, 400×). (C) Fuchsinophilic immune deposit on mesangium (MT, 400×). (D) Immune deposits are positive on C4d immunohistochemistry (inlet, 400×). (E) Electron microscopy showing mesangial increase and scattered, ill-defined electron dense deposits on mesangium (arrow) and glomerular basement membrane; subendothelial deposit (arrow head) and subepithelial deposit (serrated arrow).
GN = glomerulonephritis, PAS = periodic acid-Schiff, MT = masson trichrome.
|
Finally, the patient was diagnosed with primary, non-immunoglobulin-mediated GN. Also, we suggested complement C4d as the nature of the deposits based on the results of IHC. It is further suggested that the uveitis might be associated with the C4d-related lectin pathway. However, as retinal tissue was not available, confirmation of the cause of uveitis is not possible. The patient was treated with oral prednisolone at 20 mg per day. In four months, he showed tolerable renal function. The level of serum creatinine had not recovered but was stable. The proteinuria level remains at a similar level. On the other hand, his ocular symptoms responded to steroid therapy and the inflammatory focus was shown to have recovered on slit lamp examination.
DISCUSSION
Herein, we described a case of complement C4d-associated GN showing C4d deposition in the glomeruli with an absence of immunoglobulin or C3 deposition. The complement C4d is a split product of C4 activation by the mannose-binding lectin pathway as well as the classical pathway. In C4 glomerulopathy, bright C4d staining with no C3 or immunoglobulin deposits suggests that abnormal activation of the lectin pathway resulting from genetic factors, acquired autoantibodies, or paraproteinemia may play a role in the development of C4d deposition.5 Immune complex-mediated GN is related to the classical complement pathway, which is activated by the binding of an antibody/antigen complex to C1q, leading to the activation of C4 and generation of C3 convertase. The phenomenon is characterized by immunoglobulin, C1q, C3, and C4d staining as revealed by IF microscopy.6 C3 glomerulopathy, known as complement-mediated GN, is related to excessive activation of the alternative complement pathway through the over-activation of C3 convertase, resistance to complement factor H, and elevated levels of soluble membrane attack complex, which lead to the deposition of multiple complements in the glomeruli.27 IF microscopy showed deposits of C3 along the glomerular basement membranes, without immunoglobulin deposit. In our case, IF microscopy for immunoglobulin, C1q, and C3 did not reveal any deposits. And the glomerular deposits showed C4d IHC staining. The specimen corresponded to the newly described complement-based disease entity, C4 glomerulopathy.
Sethi group described proteinuria and hematuria as clinical manifestations of C4 glomerulopathy in their subjects.34 Those cases showed normal or slightly low C3 and C4 levels. Three patients with C4 DDD presented with proteinuria exceeding 3.5 g per day, and a patient with C4 GN had a urine protein to creatinine ratio of 430 mg/g. Two of the 4 patients showed preserved renal function, and 2 patients showed declining kidney function. In our case, the patient showed proteinuria, hematuria, and a slightly reduced C3 level and a normal C4 level. Protein excretion was not markedly increased, and SUN, and serum creatinine levels were not elevated (Table 1).
Table 1
Clinicopathological characteristics of C4 glomerulopathy

RBC = red blood cell, IFTA = interstitial fibrosis and tubular atrophy, IF = immunofluorescence, EM = electron microscopy, DDD = dense deposit disease, GN = glomerulonephritis, Cr. = creatinine, GS = global sclerosis, MPGN = membranoproliferative GN, SE = subendothelial, MES = mesangial, IN = intramembranous, TMA = thrombotic microangiopathy, NA = not applicable, MGUS = monoclonal gammopathy of undetermined significance, IHC = immunohistochemistry.
![]()
Meanwhile, our patient presented with visual disturbance, and the ophthalmologic diagnosis was uveitis. Uveitis is intraocular inflammation of the uvea, vitreous, or retina. Uveitis is categorized based on the location of the inflammatory focus and the cause of inflammation.8 The inflammatory focus was suspected to be the posterior ciliary body or vitreous by fundus photos, optical coherent tomography, and fluorescein angiography. Infection, autoimmune disease, trauma, ischemia, and cancer can cause uveitis.8 In the present case, the patient had no history of trauma, ischemia, or cancer. Hisclinical progression indicated that the inflammation was related to an autoimmune disease. Based on these factors, this case was categorized as autoimmune intermediate uveitis. Intermediate uveitis is usually an isolated eye disease but can also be a part of systemic diseases such as multiple sclerosis or sarcoidosis.8 There was no sign of systemic disease in this case. A few reports have shown that uveitis can be combined with acute nephritis or tubulointerstitial nephritis and uveitissyndrome.910 The present case showed no sign of acute interstitial nephritis. In C3 dense deposit disease, some abnormal deposits can be found on the sub-retinal pigment epithelium, which consists of electron-dense deposits in the retinal basement.11 However, there were no deposits or abnormalities on the retina or retinal pigment epithelium in this case. So, it is unclear whether the glomerulopathy and the uveitis shared the same pathogenesis, complement pathway abnormality.
In conclusion, we reported a case of C4d-dominentprimary GN. According to Sethi et al.,3 C4 glomerulopathy, including C4 GN and C4 DDD, shows characteristic features of bright C4d staining and an absence of other complement components or immunoglobulin staining by IF. C4d staining should be conducted when IF microscopic studies show negative results for immunoglobulin, C1q, and C3 in cases of electron-dense deposit-related glomerulopathy.
 XML Download
XML Download