

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Dentatorubropallidoluysian atrophy (DRPLA) is a progressive disorder characterized by a variable combination of progressive ataxia, epilepsy, myoclonus, choreoathetosis, and dementia.1 It is caused by a cytosine-adenine-guanine (CAG) expansion in the coding region of a gene on chromosome 12p13.23 In patients with DRPLA, the clinical features are related to unstable expansion of polyglutamine in atrophin-1.45
Sleep related problems, including insomnia, excessive daytime sleepiness, and rapid eye movement (REM) sleep behavior disorder (RBD), are common in neurodegenerative disease.6 However, no sleep related problems, such as RBD, have been reported in patients with DRPLA. In this report, we revealed sleep related problems in a family with DRPLA.
CASE DESCRIPTION
A 65-year-old man visited our clinic because of gait disturbance and cognitive decline in the last 15 years in 2011. He gradually developed progressive ataxia in his fifties. Although he had aggressive and violent behavior since his twenties, these behaviors had aggravated since his fifties. He had dream enactment behavior and fragmented sleep. He was physically dependent on his wife for activities of daily living. On initial neurological examination, he had slow and explosive speech. Motor power was intact, and sensory examination was normal. Knee and elbow jerks were brisk. There was generalized bradykinesia and chorea. He had dysmetria on finger-to-nose and heel-to-shin tests. His gait was slow and evidently unstable (Supplementary Video 1). On the Korean version of the mini-mental-status examination, the total score was 17 points.
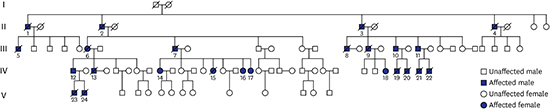
He had seven daughters. Among them, three daughters aged 31, 28, and 26 years had intractable seizures starting from adolescence (IV-15, IV-16, and IV-17, respectively; Fig. 1). These three daughters also had choreoathetosis, gait ataxia, attention deficits, and intellectual regression (Supplementary Videos 2, 3, and 4, respectively). The youngest girl had severe neurologic problems, such as gait ataxia and more frequent seizure attacks than the others. These three daughters showed dream enactment behaviors. The sixth daughter (IV-16) complained of fragmented sleep.
 | Fig. 1Pedigree from a family with DRPLA. There are 17 patients with DRPLA-presenting neurological problems such as gait ataxia, intellectual regression, and seizure.
DRPLA = Dentatorubropallidoluysian atrophy.
|
In addition, the eldest, 42-year-old daughter (IV-14), did not present any gait problem in daily life or epileptic event. However, she experienced attention deficits and a lot of emotional fluctuations since approximately 6 years ago. On neurological examination, she presented mild truncal ataxia on tandem gait (Supplementary Video 5). She complained of sleep related problems, including insomnia, highly fragmented sleep, and dream-enacting behavior. The other three healthy daughters had no clinical symptoms.
The patient's wife and healthy daughters noted that the patient's sisters also had gait disturbance, language problems, and cognitive decline. Additionally, we acquired information of family medical history from three family members to determine inheritance pattern (Fig. 1).
All the patients underwent neurocognitive evaluation, brain imaging evaluation, including brain magnetic resonance imaging (MRI) and computed tomography (CT), genetic evaluation, and overnight polysomnography (PSG). The patient who visited our clinic underwent autopsy.
First, neurocognitive evaluation showed cognitive decline by various degrees and predominantly decreased attention ability.
Second, MRI and CT showed different degrees of global cerebral atrophy and prominent atrophy in the brainstem and cerebellum (Fig. 2). Third, genetic testing revealed expansion of the CAG repeats in DRPLA (57 repeats in III-7, 58 in IV-14, 61 in IV-15, 61 in IV-16, and 63 in IV-17).
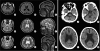 | Fig. 2Brain MRI and brain CT in patients with DRPLA. Each image shows diffuse cerebral atrophic changes with prominent atrophy of the cerebellum and brainstem in each patient IV-15 (A-C), IV-16 (D-F), and IV-17 (G-I). The most severe atrophy was observed in patient IV-17 (G-I). Diffuse cerebral atrophic changes with low densities in bilateral subcortical white matter were observed in patient III-7 (J-M).
MRI = magnetic resonance imaging, CT = computed tomography, DRPLA = dentatorubropallidoluysian atrophy.
|
Fourth, PSG evaluation showed that the patient and his symptomatic daughters presented sleep related problems, including dream-enacting behavior at night. Their sleep evaluation is summarized in Table 1. On PSG, their sleep efficiencies, that is defined as total sleep time divided by time spent in bed, were found to have significantly decreased. The Periodic Limb Movements of Sleep indices were commonly increased in these patients. All patients showed REM without atonia (Fig. 3) to various degrees or RBD. They had no sleep related respiratory problems, such as sleep apnea or hypopnea.
Table 1
Clinical Information in patients with DRPLA

DRPLA = Dentatorubropallidoluysian atrophy, CAG = cytosine-adenine-guanine, TST = total sleep time, REM = rapid eye movement, N (1, 2, 3) = Non REM Sleep stage (1, 2, 3), RDI = respiration disturbance index, PLMS = periodic limbs movement during sleep.
![]()
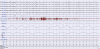 | Fig. 3Polysomnography from patient IV-17. This polysomnographic study demonstrated REM without atonia. Her chin electromyography showed increased muscle tones during REM sleep.
REM = rapid eye movement.
|
Fifth, at year 2013, 2 years from diagnosis, the patient (III-7) died from respiratory failure. Brain autopsy was performed, and the brain stem showed markedly reduced volume of the basis pontis and medulla oblongata (Fig. 4A). On coronal sectioning of the brain, the ventricles showed marked dilatation with decreased volume of the basal ganglia, thalamus, and hippocampus. On serial sections, dentate nucleus was atrophic with dark brownish discoloration. The substantia nigra and locus ceruleus showed decreased pigmentation. Microscopic examination of the cerebellar dentate nucleus showed markedly decreased numbers of neurons with reactive gliosis and the presence of 1C2-positive intranuclear inclusions (Fig. 4B). The substantia nigra showed decreased numbers of pigmented neurons in a vacuolated background with gliosis and 1C2-positive intranuclear inclusions. The remaining neurons did not show Lewy bodies, and immunohistochemical evaluation with α-synuclein antibody did not show positive inclusions. 1C2-positive intranuclear inclusions are frequently noted in the subthalamus, lateral and medial globus pallidi, caudate nucleus, thalamus, paraventricular nuclei, supraoptic nuclei, pontine neurons, oculomotor nucleus, red nucleus, and frontal cortex. Decreased numbers of neurons with gliosis was variably noted in areas with 1C2-positive intranuclear inclusions (Fig. 4C). The cerebellar cortex was relatively well preserved.

DISCUSSION
RBD is characterized by aggressive movements associated with unpleasant dreams and increased electromyographic activity during REM sleep. It has been frequently reported in trinucleotide repeat disorders and neurodegenerative diseases.789 For sleep related symptoms of patients with DRPLA, only cases involving only sleep apnea have been reported.10 Sleep related problems were not investigated in patients with DRPLA and RBD has not been reported until now.
Previous studies reported strong correlation between CAG repeat length and onset of seizure, involuntary movement, and clinical severity of the disease.11 Interestingly, the patient (III-7) and his eldest daughter (IV-14) showed RBD before presenting other neurologic symptoms. RBD was a uniformly observed initial manifestation before involuntary movement and intellectual regression in this family.
Anatomical structures that could influence REM sleep are reported in several pontinenuclei, including the noradrenergic locus coeruleus and cholinergic nuclei, pedunculo pontine nucleus, and laterodosal tegmental nucleus. In addition, forebrain structures, such as the substantia nigra, hypothalamus, thalamus, basal forebrain, and frontal cortex, are associated with REM sleep.12
Based on experimental studies in cats, the decreased activity of the coeruleus-subcoeruleus complex in the pontine tegmentum could lead to a reduction in the effect of tonic on the tegmental pedunculo pontine nucleus and magnocellular reticular formation. Magnocellular reticular formation, which has weak inhibitory effects on the spinal cord, induced tonic elevation of individual muscles. These complexes were noted in locomotor generators in the pons.13 In addition, there have been several cases of RBD associated with pontine lesions due to ischemic stroke14 and multiple sclerosis.15 In our cases, the pathological finding in the brainstem, especially in the pons, was prominent atrophic change. The structures are related to sleep related problems, including RBD; therefore, sleep related problems may develop in patients with DRPLA. However, the exact interactions of these structures in relation to REM sleep have not been clearly established.
Sleep related problems of DRPLA patients have rarely been reported, probably because of the underestimation of sleep related problems given the much higher burden of other neurological symptoms, such as seizures and involuntary movements. In addition, undergoing polysomnography for diagnosis of RBD is not easy for patients with DRPLA due to involuntary symptoms.
RBD is detectable before clinical manifestation of neuronal alpha synucleinopathy with several movement disorders. RBD has been considered a marker for some neurodegenerative disorders, such as dementia with Lewy bodies, multiple system atrophy, and Parkinson's disease.9 The preceding interval between RBD and neurodegenerative disorder has been reported as 10–20 years.16 In this family, the eldest sister (IV-14) showed initial emotional fluctuation and RBD without other definite clinical symptoms associated with DRPLA. Therefore, RBD may have a trend of manifesting into a clinical condition in this family.
Clinical manifestation may depend on the age of onset. There was no exception in these patients for the correlation between onset age and the length of CAG repeats. The symptoms in three patients (IV-15, 16, 17), with 61, 61, and 63 CAG repeats, were consistent with juvenile-type DRPLA that can show progressive myoclonic epilepsy and intellectual regression. The patient with 57 CAG repeats (III-7) was consistent with adult-type DRPLA that showed dementia and gait ataxia. The patient with 58 repeats (IV-14) had sleep related problems, including maintenance insomnia, RBD, and emotional fluctuation. Her neurologic decline progressed minimally over 4 years. At age 41 years, she did not yet show definite ataxia or cognitive decline; however, she had reported sleep and emotional problems before, similar to her father's. We are carefully observing her clinical symptoms related to DRPLA.
To our knowledge, these cases are the first reported polysomnography study of RBD in patients with DRPLA. There was not sufficient data to assess nonmotor symptoms, such as RBD. Due to this lack of data, we need additional supportive studies to determine if RBD is a clinical manifestation or a biomarker of DRPLA progression.
 XML Download
XML Download