

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Chronic obstructive pulmonary disease (COPD) is a chronic lung disorder characterized by largely not full reversible and progressive airway obstructions.12 COPD is the fourth leading cause of mortality, and will become the third by 2020.13 Though it is well known that smoking represents a major risk factor for COPD, genetic factors could also affect the development of this disease.12 To date, a large number of studies have investigated the association between genetic variations and COPD. For example, a study by Soler Artigas et al.4 reported that single nucleotide polymorphisms (SNPs) in tensin 1, the C-terminal domain of glutathione S-transferase, and in 5-hydroxytryptamine receptor 4 were associated with susceptibility to COPD. Another study by Zhou et al.5 found that several functional SNPs upstream of the hedgehog interacting protein were associated with severe COPD. Recently, several genome-wide association studies were conducted to investigate the susceptibility to COPD.6789 Based on the results of multiple meta-analyses, several SNPs that affect the Clara cell secretory protein or surfactant protein D, SNPs in cholinergic nicotinic receptor genes (CHRNA5/3), in serpin family A member 1, and in rs7937 on chromosome 19q13, show a significant association with susceptibility to COPD. However, few studies have investigated the role of genetic effects on the susceptibility to COPD in Koreans. Previous studies have reported that SNPs in interleukin-1B (IL-1B), IL-1 receptor antagonist (IL-1RA), serine peptidase inhibitor clade E2, matrix metalloproteinase-9, and CHRNA3 were associated with susceptibility to COPD in Korean populations.6101112
MER receptor tyrosine kinase (MERTK) is a member of the Axl/Mer/Tyro3 receptor tyrosine kinase family that can be activated by an endogenous ligand known as the growth-arrest-specific gene 6 (Gas6) or by protein S.1314 It is known that MERTK plays an important role in the inhibition of inflammation and in the clearance of apoptotic cells.1415 Previous studies have reported that mutations in MERTK are associated with several human diseases, such as retinal dystrophy and multiple sclerosis.16171819 Recently, it was reported that the inhibition of MERTK enhanced inflammatory responses in lipopolysaccharide-induced acute lung injuries.2021 In addition, Kazeros et al.22 found that the expression of MERTK was significantly increased in healthy cigarette smokers, compared to its expression in healthy non-smokers, and they suggested that this upregulation of MERTK might reflect the increased demand for the removal of apoptotic cells in smokers.
Here, we investigated whether genetic variations in MERTK affect the development of COPD in Koreans. We screened genomic DNA samples from 87 patients with COPD and 88 healthy controls, to identify variations in MERTK. Then, we examined the effect of each variation on the promoter activity or the expression of MERTK using in vitro assays.
METHODS
Subjects
For the case group, 87 genomic DNA samples were collected from patients with COPD who had been diagnosed by specialists in respiratory medicine at the Ewha Womans University Medical Center, or at the Kangwon National University Hospital, according to the guidelines of the Global Initiative for Chronic Obstructive Lung Disease (GOLD)23; the inclusion criteria for COPD in the present study were a post-bronchodilator ratio of forced expiratory volume in 1 second (FEV1) to forced viral capacity (FVC) of < 0.7. For the control group, 88 genomic DNA samples were collected from healthy individuals from the DNA bank of the Korea Centers for Disease Control and Prevention, Korea, or from the Kangwon National University Hospital. Demographic information such as age, sex, smoking history, and height of subjects in the control group was obtained from the Korean Genome and Epidemiology Study (4851-302) of the Korea Centers for Disease Control and Prevention, or from the Kangwon National University Hospital. Other inclusion criteria for the control group were normal findings from chest X-rays, and no history of asthma, chronic pulmonary disease, or tuberculosis.
Genetic analysis of MERTK
At first, we performed sequencing or genotyping using genomic DNA from 87 patients with COPD. To identify genetic variations in the promoter region of MERTK, a region including 2 kb upstream of the translational start site was sequenced using an automated genetic analyzer (Life Technologies Corporation, Carlsbad, CA, USA), or genotyped using the SNaPshot assay (Life Technologies Corporation). In addition, to identify genetic variations in the coding region of MERTK, the entire MERTK coding region was analyzed. Then, genotype screening for the identification of genetic variations in the promoter or coding regions in the 88 control subjects was performed using the SNaPshot assay. Haplotype assembly was performed using the Haploview software (version 4.3; Broad Institute, Cambridge, MA, USA). Nucleotide location numbers were assigned from the translational start site, based on the MERTK mRNA sequence (GenBank accession number; NM_006343.2).
Construction of plasmids containing wild-type MERTK and its variants
To construct a reporter plasmid containing the MERTK promoter region, this 1,625-bp region in MERTK was amplified from genomic DNA samples, using the NM_006343.2 reference sequence and primers that contained recognition sites for the HindIII and XhoI restriction endonucleases. The amplified products were inserted into the pGL4.11b[luc2] vector (Promega Corporation, Fitchburg, WI, USA). To construct a plasmid containing the wild-type MERTK gene, a vector (Addgene plasmid 23900) was purchased (Addgene, Cambridge, MA, USA)24 and subcloned into the pcDNA3.1 (+) vector (Life Technologies Corporation). Genetic variations in the promoter and in the coding region were obtained using the QuikChange® II Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA, USA). All DNA sequences were confirmed by direct sequencing. The primers used in this study are listed in Supplementary Table 1.
Measurement of MERTK promoter activity
Reporter plasmids containing the wild-type copy of MERTK or its variants were transfected into HCT-116 (human colon carcinoma) cells using Lipofectamine LTX and Plus reagents (Life Technologies Corporation). Thirty hours after transfection, the activity of the reporters was measured using the Dual-Luciferase® reporter assay system (Promega Corporation) according to the manufacturer's protocol, and quantified using a luminometer (Promega Corporation). The amount of transfected plasmid was normalized by using the pGL4.74 renilla vector. The firefly to renilla luciferase ratios were determined, and defined as the relative luciferase activity.
Immunoblotting
The MERTK wild-type or variation-bearing plasmids were transfected into HCT-116 cells using the Lipofectamine LTX and Plus reagents. Forty-eight hours after transfection, cells were harvested and lysed in the NP-40 cell lysis buffer, supplemented with a protease inhibitor cocktail. After centrifugation for 20 minutes at 14,000 × g at 4°C, protein concentrations were determined using the BCA assay (Thermo Fisher Scientific Inc., Waltham, MA, USA), and 50 µg of protein were loaded onto an SDS-PAGE gel. To characterize the glycosylated isoform of MERTK, 3 µL of endoH (New England Biolabs Ltd., Ontario, Canada) or 2 µL of PNGaseF (New England Biolabs Ltd.) were incubated with 10 µg of protein, and enzymatic digestion was conducted according to the manufacturer's protocol. Then, the proteins were separated on a 4%–12% SDS-PAGE gel, and transferred onto nitrocellulose membranes. The membranes were blocked for 1 hour with 5% (wt/vol) skimmed milk in Tris-buffered saline (140 mmol/L NaCl, 20 mmol/L Tris HCl, pH 7.6) and 0.1% (wt/vol) Tween-20. After blocking, the membranes were incubated with the following primary antibodies: a mouse anti-MERTK antibody (sc-365499; Santa Cruz Biotechnology, Santa Cruz, CA, USA), or a goat anti-β-actin antibody (sc-1616; Santa Cruz Biotechnology). This was followed by an incubation with the corresponding secondary antibodies in blocking buffer, and the blots were developed using the ECL detection system (GH Healthcare Life Sciences, Pittsburgh, PA, USA). The intensity of each band was measured using ImageJ (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Data analysis was conducted using the IBM SPSS Statistics software (version 23; IBM Corporation, Armonk, NY, USA). P values for the luciferase assay and immunoblotting were calculated using a one-way analysis of variance, followed by Dunnett's two-tailed test and Student's two-tailed t-test, respectively. In addition, the χ2-test was used to compare the frequency of genetic variations, or to compare the haplotypes between the case and control groups. Finally, the comparisons of demographic or clinical characteristics between the case and control groups were performed using the χ2-test for categorical variables and Student's two-tailed t-test for continuous variables. P < 0.05 was considered significant.
RESULTS
Genetic variations of MERTK in patients with COPD
Through direct sequencing or genotyping of genomic DNA from 87 patients with COPD, we identified three and seven variations in the promoter and coding regions of MERTK, respectively (Table 1). Two of the MERTK promoter variations, g.-538A>C and g.-41T>C, were first identified in this study. In the coding region, there were four nonsynonymous and three synonymous variations, and two of them, A489V and F512F, were novel.
Table 1
Frequency of MERTK genetic variations in patients with COPD

Data were obtained from DNA samples from 87 unrelated Korean patients with COPD.
MERTK = MER receptor tyrosine kinase, COPD = chronic obstructive pulmonary disease.
![]()
Comparison of the genetic variations in MERTK between the case and control groups
To compare the frequency of genetic variations in MERTK between patients with COPD and the control group, genotype screening of genetic variations in the promoter or coding regions was performed using genomic DNA from 88 healthy controls. The frequencies of the variations in MERTK in controls are listed in Supplementary Table 2. As a result, four rare variations that were found in the patient group, g.-41T>C, p.V469F, p.A489V, and p.F512F were absent in the control group. Table 2 shows the frequency of the ten variations between the two groups. We observed that there was no significant difference in the frequency of genetic variations in MERTK between the two groups. Using genotype data, haplotypes were assembled. There were three major (frequency ≥ 5%) haplotypes in our study population (Table 3). We observed that the frequency of major haplotypes in patients with COPD was comparable to that of the control group. In addition, there was no significant difference in sex, height, and smoking history between the control and case groups (Table 4). In the case of pulmonary function, the FEV1 and FEV1/FVC were much lower in the case group, when compared to those in the control group (P < 0.01).
Table 2
Frequency of MERTK genetic variations in case and control groups
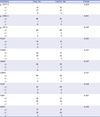
Data were obtained from DNA samples from 87 unrelated Korean patients with COPD and 88 controls. P values (+/+ vs. +/− or −/−) were obtained by comparison with control using the χ2 analysis.
MERTK = MER receptor tyrosine kinase, + = major allele, − = minor allele, COPD = chronic obstructive pulmonary disease.
![]()
Table 3
Frequency of MERTK major haplotypes in case and control groups

The minor alleles were marked in bold-faced letters with underlines.
MERTK = MER receptor tyrosine kinase.
![]()
Table 4
Demographic and clinical characteristics of subjects in case and control groups

All values are expressed as mean ± standard deviation or number (%).
FEV1 = forced expiratory volume in 1 second, FVC = forced viral capacity.
![]()
Comparison of the genetic variations in MERTK in current smokers
Because smoking represents a major risk factor for COPD, we compared the frequency of MERTK variations in current smokers only; within the COPD patient group, there were 46 smokers, while in the control group there were 56 smokers. Tables 5 and 6 show the frequencies of the variations, or those of the major haplotypes in MERTK in the two groups, respectively. The frequency of each variation in the patient group was comparable with that in the control group. However, we observed that the frequency of the wild-type haplotype (H1) was higher in the control group, although the difference was not statistically significant (P = 0.080). The smoking history was not significantly different between the two groups (Table 7). To confirm our findings for the subgroup analysis, a future analysis including a larger number of samples will be necessary.
Table 5
Frequency of MERTK genetic variations in current smokers
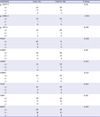
Data were obtained from DNA samples from 46 unrelated Korean patients with COPD and 56 healthy controls. All participants were smokers. P values (+/+ vs. +/− or −/−) were obtained by comparison with control using the χ2 analysis.
MERTK = MER receptor tyrosine kinase, + = major allele, − = minor allele, COPD = chronic obstructive pulmonary disease.
![]()
Table 6
Frequency of MERTK major haplotypes in current smokers

The minor alleles were marked in bold-faced letters with underlines.
MERTK = MER receptor tyrosine kinase.
![]()
Table 7
Demographic and clinical characteristics of current smokers

All values are expressed as mean ± standard deviation or number (%).
FEV1 = forced expiratory volume in 1 second, FVC = forced viral capacity.
![]()
Effects of the variations on the promoter activity of MERTK
To our knowledge, no study has investigated the function of each MERTK variation. Therefore, to characterize the functional effects of promoter variations, we constructed a reporter plasmid containing the MERTK reference sequence, and a luciferase assay was performed 30 hours after the transfection of the reporter plasmid into HCT-116 cells. As a result, the MERTK wild-type vector containing the 1,625-bp MERTK promoter region, displayed a 51-fold increase in promoter activity, compared to that in the empty vector (EV) (Fig. 1A). To examine the effect of MERTK variations on promoter activity, we constructed plasmids containing the variant sequences. After performing luciferase assays, we observed that the promoter activities of three variations, g.-1351T>C, g.-538A>C, and g.-41T>C were comparable with that of the wild-type (Fig. 1B).
 | Fig. 1Luciferase activity of wild-type MERTK and its variants. Luciferase activity measured 30 hours after the transfection of (A) the MERTK wild-type reporter plasmid or (B) reporter plasmids containing MERTK variants into HCT-116 cells. The luciferase activity of each construct is compared to that of the empty vector (EV, pGL4.11b[luc2]) (A) or the MERTK wild-type (B). The data (mean ± standard deviation) represent triplicate measurements from a representative experiment.
MERTK = MER receptor tyrosine kinase.
|
Effects of the variations on the expression of MERTK
To investigate whether variations in the MERTK coding region could affect MERTK protein expression, we performed an immunoblotting assay, following the transfection of MERTK plasmids into HCT-116 cells. As shown in Fig. 2A, MERTK was detected as two distinct bands on the western blot. We further examined the glycosylation status of MERTK after treatment with endoH or PNGaseF, followed by immunoblotting. We therefore found that the lower 140 kDa band shifted to a lower molecular weight after endoH treatment, while the upper 160 kDa band was not affected. After treatment with PNGaseF, the molecular weight of both bands had changed, suggesting that both isoforms had been deglycosylated. These data suggest that the upper band represents the fully mature, glycosylated form of MERTK, while the lower band represents the endoH-sensitive, high mannose form of MERTK. Therefore, we compared the densities of the upper bands between the wild-type MERTK and its variants, to determine the effect of variations in MERTK on its protein expression. As a result, among the seven nonsynonymous and synonymous variations, none displayed a significant difference in MERTK expression, when compared to that in the wild-type (Fig. 2B).
 | Fig. 2The effect of genetic variations on MERTK expression. (A) Immunoblotting assays performed using cell lysates obtained 48 hours after the transfection of wild-type MERTK plasmids into HCT-116 cells, in the presence of endoH (lane 2) or PNGaseF (lane 3), to examine the glycosylation status of MERTK. (B) Immunoblotting assays performed after transfection of wild-type MERTK or variant MERTK plasmids. The MERTK expression level for each variant is compared with that of the wild-type. The data (mean ± standard deviation) is obtained from three representative experiments. β-actin is used as an internal control.
MERTK = MER receptor tyrosine kinase.
|
DISCUSSION
MERTK is expressed ubiquitously on macrophages, and can be activated by Gas6 and protein S. Moreover, it negatively regulates inflammation and removes apoptotic cells after recognizing the ‘eat-me’ phosphatidylserine signal on these cells.25 The dysregulation of the immune response, or the inappropriate removal of apoptotic cells or of the debris, caused by a dysfunction in MERTK, can result in various diseases such as autoimmune diseases, chronic inflammatory diseases, and cancers.26 For example, genome-wide studies have reported the association between MERTK variations and the susceptibility to multiple sclerosis.1927 In addition, it is known that protein S, one of the ligands of MERTK presents an anti-inflammatory function, and is reduced in patients with ulcerative colitis or Crohn's disease.2829 In a mouse model, it was reported that a lupus-like disease was induced in MerTK-null mice, while another study reported that the overexpression of MerTK in mice could result in lymphoblastic leukemia/lymphoma.3031
In the airways, the clearance of apoptotic cells by MERTK is critical for the maintenance of lung homeostasis. Several studies have reported that the clearance of apoptotic immune or bronchial epithelial cells was decreased in patients with COPD, when compared to that in healthy controls.3233 In particular, this phenomenon was notably observed in smokers with COPD.33 The clearance of apoptotic cells by airway macrophages could be impaired by several factors, such as oxidative stress, that could be triggered by cigarette smoking and high mobility group protein-1.343536 Interestingly, it was observed that the expression of MERTK on airway macrophages was significantly increased in healthy cigarette smokers, compared to that in healthy non-smokers, and it was proposed that this upregulation in MERTK might reflect the increased demand for the removal of apoptotic cells in smokers in this study.22
In the present study, we hypothesized that the dysfunction in MERTK caused by genetic variations might be associated with the development of COPD. To investigate this, we screened genomic DNA samples from 87 Korean patients with COPD and from 88 healthy controls, and found that the frequencies of the variations or the haplotypes of MERTK were comparable between the two groups. Interestingly, four variations, including one promoter variation, two nonsynonymous variations, and one synonymous variation were found only in the patient group, even though the frequency of all variations was much lower in that group. In particular, three of these variations were first identified in this study. To evaluate whether these variations are COPD-specific or not, a future genetic analysis, including a larger number of samples, will be necessary. It is well accepted that cigarette smoking represents the most important risk factor for COPD; smoking induces inflammatory reactions in the airways, and suppresses the innate and adaptive immunity in the lung.37 Therefore, we compared the frequency of MERTK variations in smokers, and observed that the frequency of the wild-type haplotype was higher in the control group, although the difference was not statistically significant (P = 0.080).
To the best of our knowledge, no study has reported the effect of variations in MERTK on its gene expression. Therefore, we evaluated the effect of the MERTK variations found in our study population on MERTK promoter activity or expression, using in vitro assays. As a result, none of the variations, including three promoter variations, four nonsynonymous variations, and three synonymous variations, showed an effect on the promoter activity or the expression of MERTK.
There are several limitations in the present study. First, the number of samples was small and therefore insufficient for the results to be statistically significant. In particular, the number of smokers was extremely small. Recently, Hancock et al.38 performed a genome-wide joint meta-analysis to examine the association between genetic variations and lung function, following the investigation of SNP-by-smoking interactions. In another study, stratified genetic association analyses were conducted, according to smoking intensity, to evaluate the association between SNPs and the susceptibility to COPD.39 However, because of the small number of samples, these analyses were not performed in this study. Second, all participants in this study were of East-Asian descent. Therefore, the types and frequencies of the genetic variations could be ethnic-specific. Finally, we could not measure whether the ability of MERTK to remove apoptotic cells is affected in its variants. It is well known that the function of proteins such as enzymes, transporters, and receptors can be impaired, even when their expression remains unaffected. For example, Gautherot et al.40 reported that two nonsynonymous mutations in the multidrug resistance 3 (MDR3) transporter, encoded by the ATP-binding cassette, subfamily B, member 4 gene (ABCB4) led to a significant decrease in its transport ability, although none of these variations affected the expression of MDR3. It was subsequently found that the phosphorylation of ABCB4 was impaired by these mutations. Therefore, to clarify the effect of the MERTK variants found in our study, further functional evaluation will be required.
In conclusion, we identified ten variations in MERTK in Koreans. The frequency of each variation was comparable between patients with COPD and healthy control groups. However, in the subgroup analysis that included smokers, the frequency of the wild-type haplotype was higher in the control group, although the difference was not statistically significant. In addition, none of these variations had an effect on MERTK promoter activity or on its expression. To our knowledge, this is the first study to evaluate the association between genetic variations in MERTK and the susceptibility to COPD, along with the effect of each variation on MERTK expression by using in vitro assays. Because of the small sample size used in the present study, a further study with a larger number of samples including various ethnicities is necessary to investigate the utility of MERTK variations as predictors of COPD.
 XML Download
XML Download