

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Hemolytic uremic syndrome (HUS) is a syndromic disease characterized by thrombocytopenia, microangiopathic hemolytic anemia and acute kidney injury. Typical HUS manifests with bloody diarrhea that is mostly caused by Shiga toxin-producing enterohemorrhagic Escherichia coli infections.1 HUS is an important cause of advanced acute kidney injury in childhood, but patients generally recover renal functions completely and have a good prognosis without recurrence. HUS without bloody diarrhea (so-called atypical hemolytic uremic syndrome or aHUS) recurs often and eventually results in end-stage renal disease (ESRD) in many cases, which is different from typical HUS. In the majority of aHUS cases, complement activation via the dysregulation of the alternative pathway (AP) is a primary cause.23 Approximately 60%–70% of the genetic predispositions to aHUS are mutations in essential regulators of AP, such as complement factor H (CFH), complement factor I (CFI), membrane co-factor protein and others.456 A CFH mutation is the most common cause of aHUS. CFH is an essential regulator of AP complement activation, and the loss of function of this protein results in uncontrolled activation of AP, which induces endothelial damage and leads to thrombotic microangiopathy (TMA).
The treatment of aHUS is focused on restoring the regulation of AP to intervene in the destruction of host cells by complement activation. However, plasma therapy is often insufficient to prevent end organ damage in aHUS, and in the past approximately 70% of patients with CFH mutations died or developed ESRD within one year of presentation despite extensive plasma therapy.7 Therefore, liver transplantation (LT) has been considered as a therapeutic option for patients with defective CFH or CFI, since these factors are produced mainly in the liver.289 However, LT is a major operation with a considerable mortality rate of 8%–25%. Moreover, LT in patients with aHUS carries a higher risk of failure due to the thrombogenic tendency of the recipients.10
Eculizumab, which is a monoclonal anti-C5 antibody that blocks C5 cleavage,11 has been recommended as the first-line treatment for aHUS over plasma therapy or LT since its introduction.12 However, eculizumab is not efficacious for some causes of aHUS and is not always available due to its high price. Additionally, eculizumab therapy should be life-long, indicating that the patients will have a defective complement system for the rest of their lives. Therefore, we investigated which of the two therapies was preferable in patients with aHUS associated with a CFH mutation (CFH-aHUS).
Due to the rarity of the disease and the diversity of the clinical courses of aHUS, a prospective study comparing eculizumab and LT is not feasible; the collection of experiences with each treatment modality would be needed to obtain clinical information. Here, we present four cases of aHUS associated with CFH-aHUS. The post-kidney transplantation (KT) clinical courses were quite different according to the patients' LT statuses.
METHODS
Study design and patient collection
The cases of 4 unrelated Korean children with CFH-aHUS who were enrolled at our hospital from 1996 to 2016 were retrospectively evaluated. For this study, we reviewed the clinical and laboratory data and included patients who progressed to ESRD with CFH-aHUS and were under the age of 18 years at the time of onset.
Mutational analysis
Genomic DNA was extracted from peripheral blood nucleated cells collected from the patients and their parents. Targeted exome sequencing that covered 46 complement-related genes was performed at Samsung Genome Institute as part of the aHUS: Korean pediatric series.13 The detected mutations were confirmed with traditional Sanger sequencing.
Allograft transplantation
Allograft transplantation was performed with massive pre- and intra-operative plasma infusions to prevent thrombogenic events. Induction therapy was conducted with anti-CD20 antibodies, high-dose steroids, tacrolimus, and mycophenolate mofetil. A conventional triple regimen consisting of tacrolimus, mycophenolate mofetil and an oral steroid was given to suppress each patient's immune status after KT, and tacrolimus and/or a corticosteroid was administered after LT. The trough level of tacrolimus was 8–12 ng/mL within the first year of transplantation and then 5–8 ng/mL thereafter.14
RESULTS
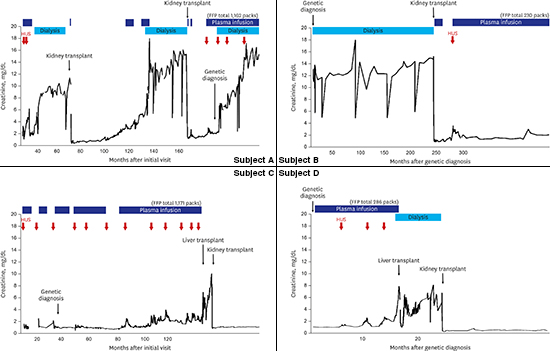
Subject A was a 20-year-old woman who was first diagnosed with aHUS at the age of 3 years. At her first presentation, she had a fever, cough and vomiting, followed by gross hematuria. The initial laboratory evaluation demonstrated severe anemia (hemoglobin, 5.1 g/dL), thrombocytopenia (platelet count, 93,000/mm3), and azotemia (creatinine, 1.8 mg/dL). Her serum albumin (3.2 g/dL), complement 3 (C3, 98 mg/dL), and complement 4 (C4, 28 mg/dL) levels were all normal (normal range: C3, 70–150 mg/dL and C4, 10–35 mg/dL). The patient was treated with a plasma infusion, plasmapheresis and hemodialysis for 6 weeks. After treatment, she recovered from acute hemolysis (hemoglobin, 9.1 g/dL and platelet count, 345,000/mm3), but the kidney injury remained (serum blood urea nitrogen [BUN], 33 mg/dL and serum creatinine, 1.4 mg/dL) and the proteinuria persisted (24-hour urine protein, 1.6 g/day). She experienced multiple episodes of aHUS, which were managed each time with plasma infusion therapy. Although she was treated properly, she progressed to ESRD at the age of 5 years and started renal replacement therapy. Three years later, she received a deceased allograft KT. Two years after the KT, her graft function started to deteriorate gradually (Fig. 1). A biopsy of the allograft kidney revealed chronic allograft nephropathy (25% global sclerosis) with occasional multilayering of the glomerular basement membrane (GBM), cellular interposition or subendothelial widening of the GBM and moderate to severe tubulitis. Since her laboratory test results were unremarkable for acute hemolysis, intravenous methylprednisolone therapy was performed repeatedly, because this situation was considered an acute rejection episode. However, her renal function did not recover. After seven months, an allograft kidney biopsy was performed again. The global sclerosis had progressed to 43%, and marked endothelial cell proliferation was noted. Because this incident was considered a recurrence of aHUS, aggressive plasma therapy was applied; however, she lost her allograft five years after her first KT. After two years of peritoneal dialysis, she received a second deceased KT; similarly, her allograft kidney function has deteriorated again in two years after the recurrence of aHUS.
 | Fig. 1Timeline of serum creatinine results and treatments for subject A.
HUS = hemolytic uremic syndrome, FFP = fresh frozen plasma.
|
Genetic analysis revealed a heterozygous mutation (c.3644G>A [p.Arg1215Gln]) of the CFH gene (Fig. 2), which was inherited from her father, and another de novo mutation (c.1649G>A [p.Cys550Tyr]) of the CFI gene. After the genetic diagnosis, the patient has been managed with regular plasma infusion therapy but is still experiencing a recurrence of aHUS.
 | Fig. 2Localization of the mutations in 4 patients. Subject A has a mutation of c.3644G>A, subject B has a mutation of c.3593A>T. Subject C has 2 mutations of c.3231T>G and c.3415C>T from both parents and subject D has a mutation of c.3572C>T.
|
Subject B was a 16-year-old girl who was first found to have ESRD at the age of 7 years. Although her medical history was uncertain, she was suspected of having aHUS because she had a history of hematuria, azotemia and decreased serum complement levels. She was transferred to our pediatric nephrology team for management of uncontrolled hypertension. On admission, her laboratory findings were hemoglobin 13.3 g/dL, a platelet count of 279,000/mm3, BUN 54 mg/dL, and creatinine 13.36 mg/dL. Her plasma C3 and C4 levels were 78 and 30 mg/dL, respectively. After a bilateral nephrectomy, her blood pressure stabilized with anti-hypertensive medications (amlodipine and irbesartan).
A genetic diagnosis was made when she was 13 years old, and a heterozygous mutation (c.3593A>T in exon 22 [p.Glu1198Val] of CFH) (Fig. 2) was found. She received a deceased donor KT at the age of 14 years. Soon after the KT, she experienced several recurrences of aHUS coinciding with infections, such as Pneumocystis pneumonia and chickenpox (Fig. 3). Although she received intensive plasma infusion therapy, her allograft renal function did not fully recover. Currently, two years after the KT, she is managed with regular plasma infusions to prevent the recurrence of aHUS, with an allograft estimated glomerular filtration rate of 39 mL/min/1.73 m2. She is still on the waiting list for a LT.
 | Fig. 3Timeline of serum creatinine results and treatments for subject B.
HUS = hemolytic uremic syndrome, FFP = fresh frozen plasma.
|
Subject C was a 12-year-old girl who presented with aHUS at one month of age with vomiting, generalized edema, and decreased urine output. At the initial presentation, she had anemia (hemoglobin, 9.3 g/dL and platelet count, 188,000/mm3), azotemia (serum BUN, 35 mg/dL and serum creatinine, 1.7 mg/dL) and hypoalbuminemia (2.6 g/dL) with proteinuria (spot urine protein creatinine ratio of 23.31). She also had low C3 (8 mg/dL) and C4 (9 mg/dL) levels. A renal biopsy revealed TMA with diffuse proliferative glomerulonephritis and C3 deposition. Her anemia and thrombocytopenia worsened (hemoglobin, 7.4 g/dL and platelets count, 77,000/mm3), and a peripheral blood smear showed schistocytes. The patient's serum CFH expression detected by western blotting was relatively low compared to that in a normal control (Fig. 4). After receiving a diagnosis of CFH-aHUS, the patient was treated with plasma infusion and recovered from acute hemolysis after four months. A genetic study of CFH confirmed the presence of a compound heterozygous mutation (c.3231T>G [p.Cys1077Trp] and c.3415C>T [p.Gln1139*]), both of which were inherited from her parents. Her renal function declined gradually as she experienced numerous episodes of aHUS that required hospital admission (45 times over a 9-year period) (Fig. 5A). When she was 9 years old, she received a deceased LT to replace the defective CFH. After the LT, her plasma complement and CFH levels normalized for the first time in her life (Fig. 4). However, the perioperative hemodynamic instability before and after surgery reduced her renal function, and she progressed to ESRD and received a living donor KT 6 months later. She is doing well without plasma infusion therapy after combined liver-kidney transplantation (CLKT) for 3 years (Fig. 5A).
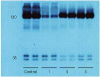 | Fig. 4Analysis of CFH binding by western blotting in subject C. The control sample is from a normal person. Lanes 1, 2, and 3 are the patient's results: lane 1 is before the LT, lane 2 is 2 weeks after the LT, and lane 3 is 25 days after the LT. The plasma CFH levels in the patient increased to within the normal range after the LT.
CFH = complement factor H, LT = liver transplantation.
|
 | Fig. 5Timeline of serum creatinine results and treatments. (A) Subject C. (B) Subject D.
HUS = hemolytic uremic syndrome, FFP = fresh frozen plasma.
|
Subject D was a 7-year-old boy who presented with aHUS at the age of 1 year. He was managed with regular plasma infusion therapy. At the age of 4 years, genetic analysis found a heterozygous mutation (c.3572C>T [p.Ser119Leu]) in CFH (Fig. 2), which was inherited from his asymptomatic mother. He was transferred to our center for a LT. On admission, his laboratory findings were hemoglobin 10.2 g/dL, a platelet count of 298,000/mm3, BUN 51 mg/dL, and creatinine 1.00 mg/dL. His plasma complement C3 and C4 levels were 112 and 36 mg/dL, respectively. He had a normal serum albumin level with proteinuria (his spot urine protein creatinine ratio was 2.1). While awaiting a deceased donor LT, his renal function gradually deteriorated with repeated episodes of recurrence, and he progressed to ESRD at 5 years old. Soon thereafter, he received a deceased donor LT and subsequently received a living donor KT from his father. After the operations, he has been free from aHUS for 14 months (Fig. 5B).
DISCUSSION
As described above, CFH-aHUS is considered a devastating disease. Patients with CFH-aHUS experience numerous episodes of recurrence and often progress to ESRD if they survive. All four cases in this study suffered from a recurrence of aHUS, usually coinciding with infection, and lost kidney functions at the age of 5–7 years. However, their presentations were diverse, probably due to their mutation types and sites, which would have different consequences on the quantity and quality of CFH. Subject C has combined heterozygous mutations in CFH; both of her CFH alleles are defective, and one allele has a stop-gain mutation leading to very low CFH and C3 levels. The other subjects, who presented later than subject C, had missense mutations on SCR20 in one of their two alleles and normal CFH and C3 levels. Although subject C suffered more frequent and more severe recurrences, her diagnosis was straightforward from a very early stage due to her very low CFH concentration; thus, the patient has been very aggressively treated with plasma. Conversely, a definitive diagnosis was uncertain for the other patients until the genetic analysis was conducted at a later time point for research purposes. Therefore, the plasma therapy applied for these patients was less aggressive than the therapy applied for subject C, which might be related to the worse outcomes. For subjects A and B, a definitive diagnosis of CFH-aHUS was obtained only after their diseases progressed to ESRD; subject A obtained her diagnosis only after she lost her first allograft (Table 1). Nonetheless, the clinical courses before the KT in the subjects were similar, with all four patients progressing to ESRD due to aHUS with episodes of acute exacerbation of renal dysfunction. However, the clinical courses after KT were quite different depending on whether the subjects had received an LT. The patients with an LT (subjects C and D) are having uneventful courses of CLKT. Despite several infection episodes, the patients without a LT (subjects A and B) have suffered from a recurrence of aHUS and repetitive insults on the allograft kidneys.
Table 1
Clinical and biochemical characteristics of CFH-aHUS patients from our study

CFH = complement factor H, aHUS = atypical hemolytic uremic syndrome, KT = kidney transplantation, DDKT = deceased donor kidney transplantation, LDKT = living donor kidney transplantation, ESRD = end-stage renal disease, LT = liver transplantation, DDLT = deceased donor liver transplantation, eGFR = estimated glomerular filtration rate.
![]()
Although plasma therapy has been the mainstay of aHUS treatment advocated by the guideline from the European Pediatric Study Group for HUS published in 2009,15 plasma therapy cannot prevent renal damage, as shown in our subjects. Additionally, plasma therapy is sometimes associated with considerable morbidity due to central line-associated bloodstream infections and a hypersensitivity to plasma.1617 Therefore, a more effective therapeutic strategy than plasma therapy is required. LT has been an option for the treatment of patients with aHUS due to defective CFH or CFI. Since 2002, approximately 25 patients who have undergone LT alone or CLKT for aHUS have been reported in the literature (Table 2).71819202122232425262728293031 The outcomes of LT have been variable, ranging from the demise of the patients from LT complications2324 to a successful long-term outcome.8921 In the current study, we report two more cases with a successful clinical course of CLKT.
Table 2
Published reports on liver or CLKT in CFH-aHUS patients

| Authors (yr) | Age at presentation | CFH mutation | Age of KT | Age of LT | Outcome |
|---|---|---|---|---|---|
| Remuzzi et al. (2002)19 | 1 yr | c.3547T>A (p.Trp1183Arg) | (+) | (+) | Alive |
| Cheong et al. (2004)20 | 3 mon | c.2777G>T (p.Cys926Phe) | (−) | 30 mon | Died |
| Remuzzi et al. (2005)21 | 13 mon | c.3514G>T (p.Glu1172*) | 3 yr | 3 yr | Died |
| Saland et al. (2006)22 | 4 mon | c.3590T>C (p.Val1197Ala) c.2918G>A (p.Cys973Tyr) compound heterozygous | 26 mon | 5 yr | Alive |
| Jalanko et al. (2008)28 | 12 mon | c.3644G>A (p.Arg1215Gln) | 18 mon | 18 mon | Alive |
| Saland et al. (2009)23 | 9 mon | c.3572C>T (p.Ser1191Leu) | 4 yr | 4 yr | Alive |
| Haller et al. (2010)29 | 5 mon | c.3644G>A (p.Arg1215Gln) | (−) | 5 yr | Alive |
| Tran et al. (2014)30 | 11 mon | c.3644G>A (p.Arg1215Gln) | 5 yr | 5 yr | Alive |
| Gonzales et al. (2016)26 | 5 mon | c.2697T>A (p.Tyr899*) homozygous | (+) | (+) | Alive |
| Coppo et al. (2016)8 | 6 mon | c.3572C>T (p.Ser1191Leu) | 5 yr | 8 yr | Alive |
CLKT = combined liver-kidney transplantation, CFH = complement factor H, aHUS = atypical hemolytic uremic syndrome, KT = kidney transplantation, LT = liver transplantation.
![]()
Eculizumab, which is a more effective therapeutic strategy than plasma therapy, currently is available in at least some parts of the world. Eculizumab therapy is a very effective treatment for aHUS considering the pathogenesis of the disease and is well established due to well-designed prospective studies. However, this therapy is very costly. In 2009, the European guidelines suggested that eculizumab was the first line therapy for aHUS.12
The long-term impairment of the immune system is a problem not only for eculizumab but also for LT, since recipients of a LT also need to take immunosuppressive medication as long as their grafted liver is functioning. Moreover, if the CFH-aHUS patient is a KT recipient, the patient needs to take immunosuppressive medications to prevent rejection of the KT even when the patient is managed with eculizumab. Thus, for KT recipients, LT would not result in additional immunosuppression. In the past, the surgical outcome of LT was not satisfactory for aHUS patients due to their thrombogenic diathesis, but recently the application of perioperative plasmapheresis or eculizumab therapy has much improved the prognosis of LT in CFH-aHUS patients.24 Therefore, for KT recipients with CFH-aHUS, LT may be a better choice than eculizumab; for instance, our subjects C and D, who are CLKT recipients, will not require eculizumab if their liver allografts last.
However, LT is only applicable for patients with proven causative mutations of CFH or CFI whose main source is the liver.18 Patients with mutations of other compliment regulators or whose mutations have not been found are not eligible for LT, because LT may not satisfactorily restore the defective component. Conversely, non-responders of eculizumab would have to choose to receive an LT if they had defective CFH or CFI.
In the past, isolated LT was considered early in the course of CFH-aHUS to prevent further damage of the kidneys and to preserve renal functions.23 Is this approach still feasible in this era of eculizumab? From a medical cost perspective, the answer is yes. From the immunological perspective, which option is a better choice is unknown. LT has its own limitations (i.e., it may be rejected at any time and is not reversible), whereas eculizumab may be discontinued at any time if indicated. Moreover, the clinical courses of CFH-aHUS are quite variable.2326 Although all of the patients described in this study lost their kidneys, some patients with the same genetic background grow out of the disease and do not need a constant restoration of CFH or the inhibition of complement activation; for example, some of the parents of our subjects were completely asymptomatic even though they had same CFH mutation as the subjects. Therefore, a genetic diagnosis of CFH-aHUS per se is not an indication for LT32333435 even if the procedure is feasible. For patients with CFH-aHUS whose course is not devastating, eculizumab is the better choice, because it can be discontinued when the patient's condition improves.
Additional studies are needed to evaluate the long-term prognosis and risk-benefit of LT or CLKT. Additionally, clinicians should deeply consider the best treatment for refractory aHUS and maintain their adherence to national or international registries for aHUS patients after transplantation.24
For patients with CFH-aHUS, plasma therapy is not sufficiently effective to prevent renal failure. In CFH-aHUS patients, KT without LT was complicated with the recurrence of aHUS, which might lead to allograft loss. Conversely, LT was successful for the prevention of the recurrence of aHUS. LT may be an option for an aHUS-free life for aHUS patients with proven genetic defects in CFH or CFI, even in the era of eculizumab, which is a costly standard therapy.
 XML Download
XML Download