

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Occupational exposure accounts for 17% of adult-onset asthma.1 Toluene diisocyanate (TDI) is a commonly used diisocyanate and is known to be a cause of occupational asthma (OA).2 Subjects with TDI-induced OA (TDI-OA) usually experience progressive declines in lung function and sustained respiratory symptoms even in the absence of additional exposure.3 Exposure to TDI can occur in workplaces that produce flexible polyurethane foam and other products.4 Although the incidence of TDI-OA has been declining through protection and regulation policies, use of TDI has not decreased and many workers are still at high risk of TDI-OA.5 TDI has a low molecular weight and is a highly reactive chemical.6 Although many efforts to address the pathogenesis of TDI-OA have been made, its nature is heterogeneous in which various immunologic and non-immunologic mechanisms are involved.
Periostin is a secreted, 90-kDa extracellular protein with a structure homologous to adhesion molecule fasciclin I.7 It has the ability to bind fibronectin, tenascin-c, and collagen and to constitute subepithelial fibrosis with other extracellular matrix proteins.8 Periostin plays an important role in inflammation as a matricellular protein.9 It has autocrine effects in bronchial epithelial cells and paracrine effects in airway fibroblasts to activate transforming growth factor-β1 (TGF-β1) and type I collagen.10 Periostin has been implicated in various allergic diseases, such as asthma, chronic rhinosinusitis with nasal polyp, allergic conjunctivitis, and atopic dermatitis.1112 However, the role of periostin in the pathogenesis of TDI-OA has not yet been completely elucidated.
As periostin may be involved in the pathogenic mechanisms of airway inflammation and remodeling in TDI-OA, this study was conducted to compare serum periostin levels among TDI-OA subjects, asymptomatic TDI-exposure controls (AECs), adult non-occupational asthmatics (NAs), and normal controls (NCs) not exposed to TDI and to investigate underlying mechanisms by which periostin is released from airway epithelial cells in TDI-exposed workers.
MATERIALS AND METHODS
Study population
Fifty-three subjects with TDI-OA, 71 AECs, 67 NAs, and 83 NCs were recruited in Ajou University Medical Center (Suwon, South Korea). TDI-OA was defined as the presence of a history of TDI exposure in the workplace and a positive response to TDI provocation test. AECs were subjects with a history of exposure to TDI in the workplace and no symptoms relating due to asthma. NAs were subjects diagnosed with aspirin tolerant and non-severe asthma. NAs or NCs did not have a history of exposure to TDI. Blood samples were collected 1 week after discontinuation of all asthma medications, including inhaled/systemic corticosteroids and leukotriene receptor antagonists, and were stored at −20℃ until used. Blood sampling was performed at the initial visit of each patient at which time most of the study subjects had been exposed to TDI. Atopic status was determined by results of skin prick test with common inhalant allergens (Bencard, Bradford, UK), wherein atopy was considered present if the test elicited positive reactions to at least one allergen. Total IgE was measured using the ImmunoCAP system (Thermo Fisher Scientific, Waltham, MA, USA). Informed consent was obtained from all patients. This study was approved by the Ethical Review Board of Ajou University Hospital (AJIRB-GENSMP-13-108).
Provocation test with methacholine and TDI
Methacholine bronchial provocation test was conducted according to generally accepted guidelines.13 Doubled doses of methacholine (0.075 mg/mL to 25 mg/mL) were inhaled by the dosimeter method at 5 minute intervals until a fall in forced expiratory volume in 1 s (FEV1) 20% or more. PC20 level, a concentration of methacholine in which FEV1 decrease by 20%, was calculated by interpolation from the dose-response curve between methacholine and FEV1. TDI provocation test was performed following a previously described method.14 Subjects were exposed to TDI (2,4 form:2,6 form=80:20; Sigma-Aldrich, St Louis, MO, USA) for 5 to 45 minutes through tidal breathing in a closed room until respiratory symptoms occurred. FEV1 was measured before and 5 hours after the TDI exposure. If a fall in FEV1 was 20% or more, the results of TDI provocation test was considered positive.
TDI-HSA conjugate
TDI-human serum albumin (TDI-HSA) conjugate was kindly provided by Dr. Justin M. Hettick (National Institute for Occupational Safety and Health, Morgantown, WV, USA), and 2,6-TDI was conjugated by HSA at different ratios and at a concentration of 0.5 mg/mL. Three different ratio (TDI:HSA), 40:1, 10:1 and 5:1, were provided. Conjugation between TDI and HSA, cross-linking, and reactivity were demonstrated.15 Among them, TDI-conjugated HSA at 40:1 was more reactive than the others in enzyme-linked immunosorbent assay (ELISA). Therefore, 40:1 TDI-HSA conjugate was used in this study.
Measurement of periostin and other cytokines
Periostin, interleukin-8 (IL-8), and TGF-β1 levels in serum samples and cell culture supernatants were measured by commercial ELISA kits (Shino-Test Corporation, Tokyo, Japan; Endogen, Woburn, MA, USA; and R&D Systems, Minneapolis, MN, USA, respectively), following the manufacturers' instructions. The serum levels of myeloperoxidase (MPO) and IL-1β in cell supernatants were also measured by commercial ELISA kits (Biocheck Inc, Foster City, CA, USA and R&D Systems, respectively).
Measurement of intracellular reactive oxygen species
2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) was purchased from Molecular Probes (Eugene, OR, USA) and was used as a fluorescent probe to detect the extent of reactive oxygen species (ROS) activation. Isolated neutrophils from asthmatic patients at a concentration of 1×105 cells/mL were treated with phosphate buffered saline (PBS), TDI-HSA conjugate, and TDI-HSA conjugate with N-Acetyl-L-cysteine (NAC) (Sigma-Aldrich) for 24 hours and incubated with 50 µM H2DCFDA for 30 minutes at 37℃. They were analyzed using a FACS Canto II flow cytometer (BD Biosciences, San Jose, CA, USA).
Isolation of peripheral blood neutrophils
Venous blood was collected from adult asthmatics in BD Vacutainer tubes containing acid citrate dextrose solution (BD Biosciences) and was processed immediately after collection following a previously described method.16 Blood was layered on Lymphoprep solution (Axis-Shield, Oslo, Norway), which was then centrifuged at 2000 rpm and 20℃ for 25 minutes. The bottom red layer was mixed with Hank's balanced salt solution (HBSS) buffer containing 2% dextran (Polysciences, Warrington, PA, USA) and was maintained for 30 minutes at room temperature. The upper layer containing neutrophils was collected, followed by centrifugation at 300 g and 20℃ for 8 minutes with HBSS buffer. The remaining red blood cells were removed by hypotonic lysis. Isolated neutrophils were maintained in RPMI-1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 2% fetal bovine serum (FBS). Cell purity was determined by hematoxylin and eosin staining or flow cytometry.
Human airway epithelial cell culture and treatment
Three human airway epithelial cells (HAECs), including A549 cells, BEAS-2B cells, and primary small airway epithelial cells (SAECs), were purchased from American type Culture Collection (ATCC, Manassas, VA, USA) and were cultured following previously reported methods.17 A549 and BEAS-2B cells were cultured in RPMI-1640 medium with 10% heat-inactivated FBS, penicillin (100 IU/mL), and streptomycin (50 µg/mL). SAECs were cultured in Airway Epithelial Cell Basal Medium (ATCC), supplemented with HSA 500 mg/mL, linoleic acid 0.6 mM, lecithin 0.6 mg/mL, L-glutamine 6 mM, extract P 0.4%, epinephrine 1.0 mM, transferrin 5 mg/mL, triiodothyronine 10 nM, hydrocortisone 5 mg/mL, epidermal growth factors 5 ng/mL, and insulin 5 mg/mL. Cells were maintained at 37℃ with 5% CO2 in humidified air.
HAECs (1×105 cells/mL) were treated with 10 µL of 1 µg/mL TDI-HSA conjugate and/or 10 µL of 1 mM NAC for 24 hours. Isolated neutrophils at 0.5×105 cells/mL, an optimal count in previous experiments, were co-cultured with HAECs in transwells to interfere with direct contact between neutrophils and HAECs. Supernatants were collected and frozen at −80℃ until ready for use.
Statistical analysis
Subject characteristics were analyzed using Student's t test or the Mann-Whitney U test for continuous variables and the chi-square test or Fisher's exact test for categorical variables. Analyses among three or more groups were performed using one-way ANOVA with Bonferroni post hoc test. Correlations between periostin and other cytokines were analyzed by applying Spearman's rank coefficient (r). Receiver operating curves (ROCs) were used to determine a cutoff value for serum periostin levels that differentiated TDI-OA and TDI-exposed subjects from healthy controls. Clinical and inflammatory parameters were compared by a generalized linear model between subjects with a higher periostin level and those with a lower periostin level. All statistical analyses were performed with SPSS version 23.0 (IBM Corp., Armonk, NY, USA). A p value of less than 0.05 was considered statistically significant.
RESULTS
Characteristics of the study subjects
The mean ages of the subjects with TDI-OA, AECs, NAs, and NCs were 42.8±9.6, 40.0±9.0, 46.5±12.5, and 39.1±13.5 years, respectively (p<0.001) (Table 1). The proportions of males among subjects with TDI-OA, AECs, NAs, and NCs were 64.2, 69.0, 44.8, and 53.6%, respectively (p=0.001). The proportion of ever smokers among AECs (59.4%) was significantly higher than those among TDI-OA, NAs, and NCs (25.6, 14.3, and 8.4%, respectively, p<0.001). The duration of TDI exposure was significantly shorter in subjects with TDI-OA than in AECs (6.9±4.6 vs. 13.3±8.6 years, p=0.015). The serum total IgE levels, FEV1 (%), and PC20 levels were comparable among the groups.
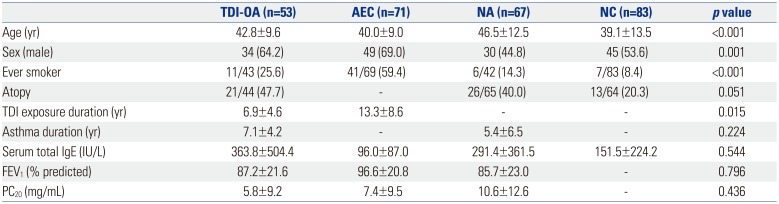
Table 1
Clinical Characteristics of the Study Subjects
TDI, toluene diisocyanate;TDI-OA, TDI-induced occupational asthma; AEC, asymptomatic TDI-exposure control; NA, non-occupational asthma; NC, normal healthy control; FEV1, forced expiratory volume in 1 s; PC20, concentration of methacholine required to decrease a 20% in FEV1.
Data are presented as the mean±SD or n (%).
![]()
Elevated levels of serum periostin in TDI-OA
The mean serum periostin level was 127.6±79.5 ng/mL in subjects with TDI-OA, which was significantly higher than that in AECs, NAs, and NCs (100.9±33.5, 75.3±34.6, and 58.7±29.8 ng/mL, respectively, all p<0.05) as shown in Fig. 1.
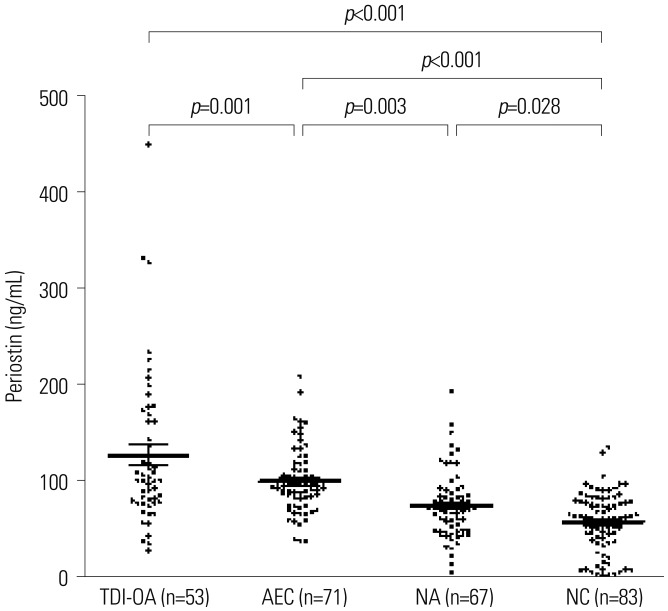
Fig. 1
Comparison of serum periostin levels in the four study groups (subjects with TDI-OA, AEC, subjects with NA, and NCs). Periostin levels were measured using ELISA. Significantly increased serum periostin levels were noted in TDI-OA, compared to AEC, NA, and NC. TDI, toluene diisocyanate; TDI-OA, TDI-induced occupational asthma; AEC, asymptomatic TDI-exposure control; NA, non-occupational asthma; NC, healthy normal control. Results are presented as means±SEM.
![]()
Comparison of clinical and immunologic parameters in subjects with TDI-OA plus AECs
Clinical and immunologic parameters in subjects with TDI-OA plus AECs were analyzed according to a cutoff periostin value of 116.7 ng/mL, which was calculated from the mean plus 2 standard deviations of the serum periostin level in NCs (Table 2). Subjects with a lower periostin level were predominantly male (75.9% vs. 45.9%, p=0.002) and ever smokers (55.6% vs. 22.6%, p=0.003), compared to those with a higher periostin level. The PC20 methacholine levels were significantly lower in subjects with a higher periostin level than in those with a lower periostin level (1.9±2.5 mg/mL vs. 8.0±10.5 mg/mL, p=0.028). The serum levels of IL-8, MPO, and TGF-β1 were not significantly different between the two groups. Serum periostin levels were examined for their correlations with IL-8, MPO, and TGF-β1 levels. No significant correlations were found between periostin and cytokines measured: IL-8 (r=0.135, p=0.122), MPO (r=−0.042, p=0.679), and TGF-β1 (r=−0.032, p=0.761).
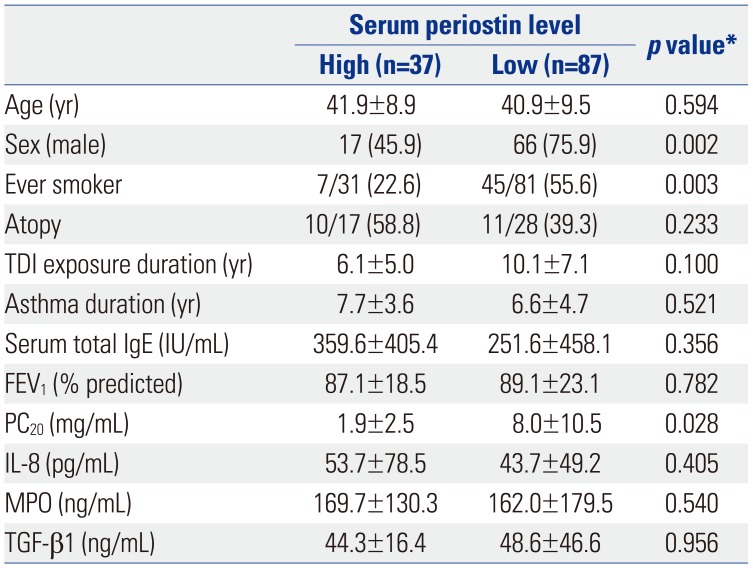
Table 2
Comparison of Clinical and Immunologic Parameters in Terms of Serum Periostin Level in TDI-Exposed Subjects
![]()
The serum periostin level at which AECs and subjects with TDI-OA were differentiated from NCs was examined using ROC analysis. AECs and subjects with TDI-OA were well discriminated from NCs at a periostin level of 81.5 ng/mL with area under the curve (AUC) of 0.839, 74.2% sensitivity and 78.4% specificity (p<0.001) (Supplementary Fig. 1A, only online). Subjects with TDI-OA were discriminated from NCs at a cutoff periostin value of 89.5 ng/mL with AUC of 0.837, 64.2% sensitivity and 87.5% specificity (p<0.001) (Supplementary Fig. 1B, only online). However, TDI-OA could not be discriminated from AEC in ROC analysis with a serum periostin (AUC of 0.578, p=0.126).
ROS production by neutrophils in response to TDI-HSA conjugate
In asthmatic patients, ROS production by neutrophils in response to TDI-HSA conjugate was measured by mean fluorescence intensity of fluorescent probe H2DCFDA using flow cytometry. ROS production by neutrophils was significantly increased after treatment with TDI-HSA conjugate, compared to treatment with PBS (Fig. 2A). However, NAC suppressed ROS production to a similar extent as that with PBS. No significant effect of TDI-HSA conjugate on IL-1β production by neutrophils was noted (Fig. 2B).
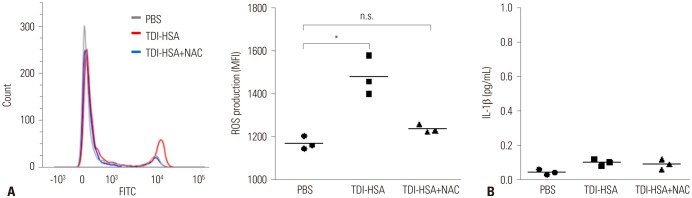
Fig. 2
TDI-HSA conjugate-induced neutrophil activation according to ROS production. (A) Flow cytometry analysis of ROS production measured by the MFI of fluorescent probe H2DCFDA. (B) There was no significant effect of TDI-HSA conjugate on the production of IL-1β by neutrophils. *p<0.01 obtained by one-way ANOVA with Bonferroni post hoc test. TDI, toluene diisocyanate; PBS, phosphate buffered saline; TDI-HSA, TDI-human serum albumin; NAC, N-Acetyl-L-cysteine; ROS, reactive oxygen species; MFI, mean fluorescence intensity; H2DCFDA, 2′,7′-dichlorodihydrofluorescein diacetate; n.s., not significant.
![]()
Effect of TDI and neutrophils on immune response in HAECs
Three kinds of HAECs were co-cultured with TDI-HSA conjugate. Their viability was evaluated by CCK8 assay. No significant change in viability was detected (Supplementary Fig. 2A, only online). In addition, the release of IL-8 and IL-6 from HAECs was not affected by treatment with TDI-HSA conjugate (Supplementary Fig. 2B and C, only online).
IL-8 release was significantly increased after co-culture with neutrophils in BEAS-2B cells and SAECs in response to TDI-HSA conjugate (Fig. 3A). Periostin was not detected in cell supernatants from A549 and BEAS-2B cells regardless of neutrophil co-culture and TDI-HSA treatment. In SAECs, periostin was released at a low level without any treatment. Although TDI-HSA conjugate alone did not increase periostin, periostin increased significantly when co-cultured with neutrophils, which was further increased by treatment with TDI-HSA conjugate. NAC decreased periostin to a similar extent as that in negative controls (Fig. 3B). In addition, co-culture with neutrophils significantly increased TGF-β1 from SAECs, which was further increased by treatment with TDI-HSA. However, NAC remarkably decreased TGF-β1 (Fig. 3C). In addition, within culture supernatants from SAECs, periostin levels were significantly correlated with IL-8 levels (r=0.941, p<0.001) (Fig. 3D), and TGF-β1 levels were significantly correlated with periostin (r=0.830, p<0.001) (Fig. 3E) and IL-8 levels (r=0.829, p<0.001) (Fig. 3F).
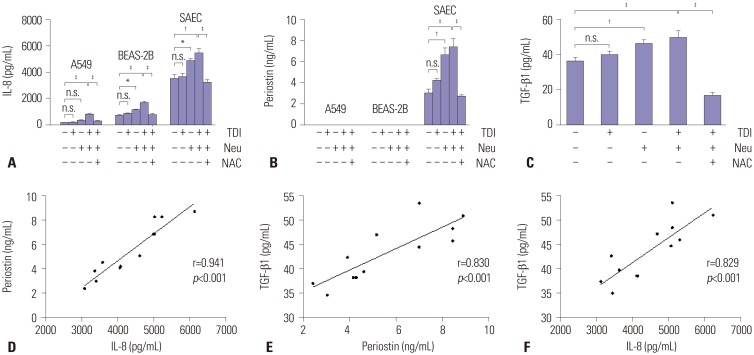
Fig. 3
Effects of TDI-HSA conjugate on the induction of immune responses by neutrophils in airway epithelial cells. Concentrations of (A) IL-8, (B) periostin, and (C) TGF-β1 released from airway epithelial cells in the presence of neutrophils under trans-well. Correlation of periostin with (D) IL-8 and TGF-β1, as well as with (E) periostin and (F) IL-8. *p<0.05, †p<0.01, ‡p<0.001 obtained one-way ANOVA with Bonferroni post hoc test. TDI, toluene diisocyanate; TDI-HSA, TDI-human serum albumin; SAEC, small airway epithelial cell; NAC, N-Acetyl-L-cysteine; n.s., not significant; Neu, neutrophils.
![]()
DISCUSSION
In the present study, we found that serum periostin levels were significantly higher in TDI-OA than in AECs, NAs, and NCs. PC20 methacholine levels were significantly lower in subjects with a higher periostin level than in those with a lower periostin level. TDI exposure did not increase periostin production directly by airway epithelial cells. However, periostin production by airway epithelial cells increased significantly after coculture with neutrophils, which were suppressed by an antioxidant. In addition, TGF-β1 production by airway epithelial cells increased when exposed to TDI and neutrophils, which was also suppressed by an antioxidant. This is the first study to suggest that high periostin levels induced by airway epithelial cells may contribute to the progression of airway inflammation leading to airway remodeling in subjects with TDI-OA.
Serum periostin has been regarded as a biomarker for bronchial asthma, especially in T-helper 2 (Th2)-driven asthma phenotype.18 IL-13 is a central cytokine for the development of type 2 immune response.19 The periostin gene POSTN is highly expressed by IL-13 in bronchial epithelial cells.20 In immunohistochemical analysis of bronchial biopsy specimens from asthmatic patients, periostin was deposited on the thickened basement membrane, indicating that periostin plays a pathogenic role in bronchial asthma as a constituent of subepithelial fibrosis.8 Gene expression studies with airway epithelial brushings and sputum cells have shown that periostin is also found to be highly expressed in the Th2-high subtype, compared to the Th2-low subtype.2122 In this regard, biologic agents targeting type 2 immunity, such as anti-IL-13 antibody (lebrikizumab) and anti-IgE antibody (omalizumab), may have a significant therapeutic effect in subjects with higher periostin levels, compared to those with lower levels.2324 However, despite accumulating evidence of periostin involvement in Th2 phenotype of bronchial asthma, there is a lack of research on whether periostin is associated with other phenotypes of asthma.
In the present study, the mean serum level of periostin in subjects with TDI-OA was 127.6 ng/mL, which is higher than levels in other study subjects with non-occupational asthma (ranging from 76.5 ng/mL to 92.8 ng/mL).2526 These findings suggest that another mechanism other than type 2 immunity may contribute to the production of periostin by airway epithelial cells. Various immunologic and non-immunologic mechanisms have been shown to be involved in the pathogenesis of TDI-OA. Among them, ROS production in response to TDI exposure in the workplace has been found to play a crucial role in the development of airway inflammation.27 In addition, exposure to TDI rapidly decreases glutathione levels,28 and other antioxidants, such as thioredoxin, ferritin, and heme oxygenase-1, are also affected in TDI-OA.29 As a result, ROS can initiate inflammatory responses through activation of nuclear factor-kappaB and hypoxia-inducible factor, which induce inflammatory cascades in airway epithelial cells.3031 Moreover, production of IL-8, a potent chemotactic factor for neutrophils, is increased in airway epithelial cells in response to TDI.32 Activated neutrophils are the most intense producers of ROS.33 In the present study, TDI increased ROS production by neutrophils, and neutrophils increased release of periostin from epithelial cells, which was further increased by TDI and suppressed by NAC, an antioxidant. Therefore, increased ROS production by neutrophils can be considered another contributing factor to the release of periostin from airway epithelial cells in pathogenic mechanisms of airway inflammation of TDI-OA.
TGF-β1 is considered a key mediator of airway remodeling including subepithelial fibrosis.34 Airway remodeling is closely linked with the degree of airway hyperresponsiveness.35 Activated airway epithelial cells release a large amount of periostin, which in turn activates TGF-β1 production by epithelial cells through an autocrine effect mediated by matrix metalloproteinase (MMP)-9.36 MMP-9 was also found to be increased in induced sputum from subjects with TDI-OA after the TDI provocation test.37 In the present study, subjects with a higher periostin level showed more severe airway hyperresponsiveness to methacholine than those with a lower periostin level. Additionally, periostin release from SAECs was well correlated with that of TGF-β1. Although whether TGF-β1 contributes to airway remodeling was not confirmed in this study, these experimental results indicate ROS-induced periostin released from airway epithelial cells may interact with TGF-β1.
There is no direct evidence to demonstrate that periostin is associated with neutrophilic inflammation. However, recent studies on idiopathic pulmonary fibrosis (IPF) suggest that periostin is a potential mediator involved in airway neutrophilic inflammation. Serum periostin levels were found to be significantly higher in patients with IPF than in those with other subtypes of interstitial lung disease, and lung function was inversely correlated with periostin levels.38 In a mouse model of bleomycin-induced IPF, pulmonary fibrosis, recruitment of neutrophils and chemokines for neutrophils were significantly decreased in periostin-deficient mice, suggesting that periostin may be associated with neutrophil activation in lung tissue.39 TDI-OA has been regarded as a phenotype of severe asthma with airway remodeling and is characterized by neutrophilic inflammation.27 In the present study, neutrophils increased periostin release from airway epithelial cells, which was further increased after co-exposure to TDI and neutrophils. These findings suggest that TDI exposure, along with neutrophilic inflammation, as well as Th2 immune responses, could increase periostin production by airway epithelial cells, thereby exacerbating the progression of airway inflammation and remodeling, resulting in a decline in lung function in subjects with TDI-OA.
Several studies have outlined the roles of epithelial cell-derived molecules (folliculin, clusterin, and progranulin), which are mediated by ROS injury, in the pathogenesis of TDIOA.4041 They can be induced from airway epithelial cells under short-term exposure to TDI with neutrophils in in vitro systems, while clusterin/progranulin may be depleted with chronic exposure. Therefore, lower serum levels of clusterin/progranulin were noted in TDI-OA patients, which may enhance ROS injury. The present study suggests that periostin is an additional epithelial cell-derived molecule in response to TDI/neutrophils-induced ROS. Future research needs to study interactions among these mediators in the pathogenesis of TDI-OA.
Periostin has been suggested as a potential biomarker for chronic upper and lower airway diseases, including Th2-asthma.42 Periostin is easily secreted from inflamed tissues into body fluids, including blood and sputum.4344 Serum periostin levels can appropriately reflect inflamed tissue and can be more accurately detected, compared to other biomarkers. In the diagnosis of TDI-OA, there have been no studies to demonstrate and validate a reliable in vitro test to differentiate TDI-OA subjects from those with the other types of non-occupational asthma or AEC and NC individuals. In this regard, the present study suggests that a serum periostin level higher than a specific cut-off value may be a potential serum biomarker for predicting the phenotype of TDI-OA with high diagnostic accuracy, although more prospective studies in other cohorts of TDI-exposed workers will be needed to validate this hypothesis. Also, further studies will be needed to explore the exact mechanism by which periostin induces TGF-β1 production in the airway mucosa of subjects with TDI-OA and whether a high periostin level can be used to predict progression of airway remodeling in subjects with TDI-OA.
There are several limitations in this study. First, we collected neutrophils isolated from non-occupational asthmatic patients, which may not be able to reflect exact functional status of neutrophils derived from subjects with TDI-OA. Secondly, we did not demonstrate that ROS directly mediated production of periostin and TGF-β1 by SAEC. It is possible that other mediators released from TDI-HSA activated neutrophils promote production of them. Thirdly, the long-term prognosis of subjects with a higher periostin level, especially AECs, was not evaluated, since this study was conducted as a cross-sectional study. A long-term patient cohort is warranted.
In conclusion, the results of this study suggest that increased levels of periostin may contribute to progression of airway inflammation/remodeling in the airway mucosa of subjects with TDI-OA, leading to poor outcomes. Serum levels of periostin could be a potential serologic marker for representing the phenotypes of TDI-OA. Further prospective studies are needed to monitor the long-term prognosis of subjects with TDI-OA stratified by serum periostin levels.
 XML Download
XML Download