

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Mitochondrial disease is a complex and heterogeneous multisystem disorder that is caused by mitochondrial dysfunction, particularly defects in the mitochondrial respiratory chain (MRC) complex and related aberrant oxidative phosphorylation.1 Mitochondrial disease has dual origins: it may develop because of deformities in mitochondrial DNA (mtDNA) or nuclear DNA at the genetic level.2 These conditions can cause numerous phenotypes and genotypes of mitochondrial diseases, leading to the involvement of multiple organs, with variations in the extent and severity of the manifestations, as well as different clinical outcomes.3 Because of this complexity, mitochondrial disease may present with any symptom in any organ at any age.4
Mitochondrial diseases that involve ocular symptoms as a major manifestation include chronic progressive external ophthalmoplegia (CPEO) and Kearns-Sayre syndrome (KSS), among others.5 CPEO, a comparably mild mitochondrial disease, is characterized by weakness in the extraocular muscles and ptosis, and it is often accompanied by limb weakness.6 Ptosis typically progresses to ophthalmoparesis over months or years.7 KSS is defined as CPEO that begins before 20 years of age, is accompanied by pigmentary retinopathy, and is associated with at least one of the following: cardiac conduction deficits, cerebellar ataxia, or an elevated cerebrospinal fluid protein concentration.6
Mitochondrial diseases with ophthalmoplegia, such as KSS, are very rare, and the natural courses of these types of diseases are not well known. Therefore, in this study, we systematically reviewed the clinical characteristics, diagnosis, and natural courses of mitochondrial diseases with ophthalmoplegia.6
MATERIALS AND METHODS
Patients and study design
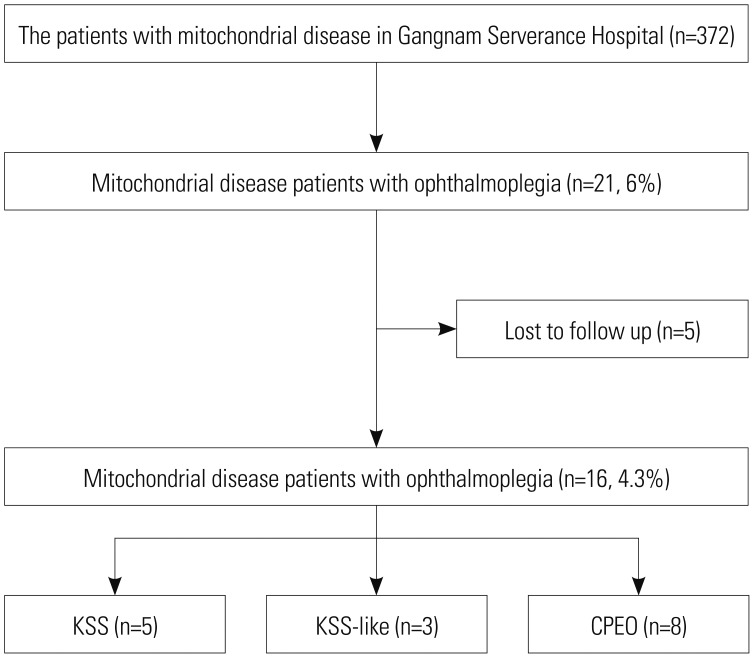
We retrospectively reviewed the medical records of 372 patients with mitochondrial disease who were followed up regularly at our hospital between January 2006 and January 2016 (Fig. 1). We identified 21 patients with ophthalmoplegia symptoms out of the 372 patients with mitochondrial disease, although five patients who were lost to follow-up were excluded. The final study population included 16 patients with mitochondrial disease and symptoms of ophthalmoplegia. All patients satisfied the modified criteria for mitochondrial disease proposed by Bernier, et al.,8 which include clinical, histopathological, enzymatic, and metabolic parameters. The patients were classified into three groups: KSS, KSS-like, and CPEO. KSS is defined as CPEO that begins before 20 years of age and is accompanied by pigmentary retinopathy and cardiac conduction disease. Patients were placed in the KSS-like group if they had KSS without cardiac conduction disease. The other patients were placed in the CPEO group, regardless of age. Clinical severity was defined as follows: mild, ambulatory and/or independent for daily activities; moderate, wheelchair-bound and/or partially dependent for daily activities; severe, bedridden and totally dependent for daily activities; and deceased. The study was approved by the Institutional Review Board of the Yonsei University Gangnam Severance Hospital (3-2017-0168). Informed consent was obtained from parents or legal guardians for all diagnostic procedures and molecular studies.
Collection of data regarding clinical characteristics and diagnostic evaluation
Disease-related clinical variables were collected, including the type of the first symptom, family history, age of onset of ocular myopathy, age of onset of the first symptom, duration from the first symptom to the last outpatient visit, and clinical severity at the last outpatient visit. Muscle biopsy was performed, and the samples were assessed via routine histology, immunohistochemistry, and electron microscopy examinations.9
We analyzed diagnostic data obtained from histopathological examinations, enzymatic assays for MRC, laboratory tests, neuroimaging studies, and a gene study. We used a strategy based on long PCR for analysis of mtDNA from the patients. PCR primer sets were designed to cover the KSS common deletion mtDNA 5-kb and 7.4-kb deletion breakpoint. All PCR products were directly sequenced in both directions.
Syndromic diagnosis was performed. If a patient's serum lactate level was at least two times the normal value, the level was considered as severely increased; serum lactate levels above normal (0.5–2.2 mmol/L), but less than two times the normal value, were considered as mildly increased. Serum creatine kinase levels were classified in a similar manner.
Statistical analysis
All statistical analyses were conducted using Statistical Package for the Social Sciences (SPSS), version 20.0 (IBM Corp., Armonk, NY, USA). The data are expressed as medians and interquartile ranges. Because our study population was small, the KSS and KSS-like groups were combined and compared with the CPEO group. Fischer's exact tests and chi-square tests were used to evaluate differences between the groups. Mann-Whitney U tests were used to analyze the duration-related variables. p values less than 0.05 were considered statistically significant.
RESULTS
Patient characteristics
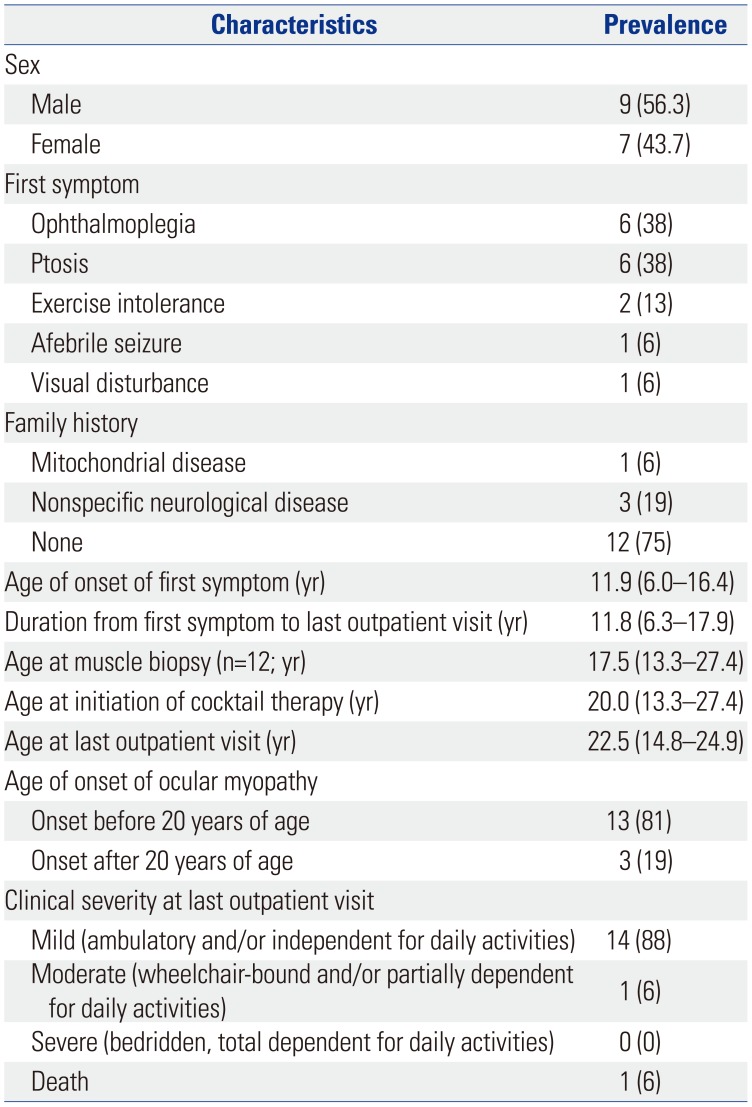
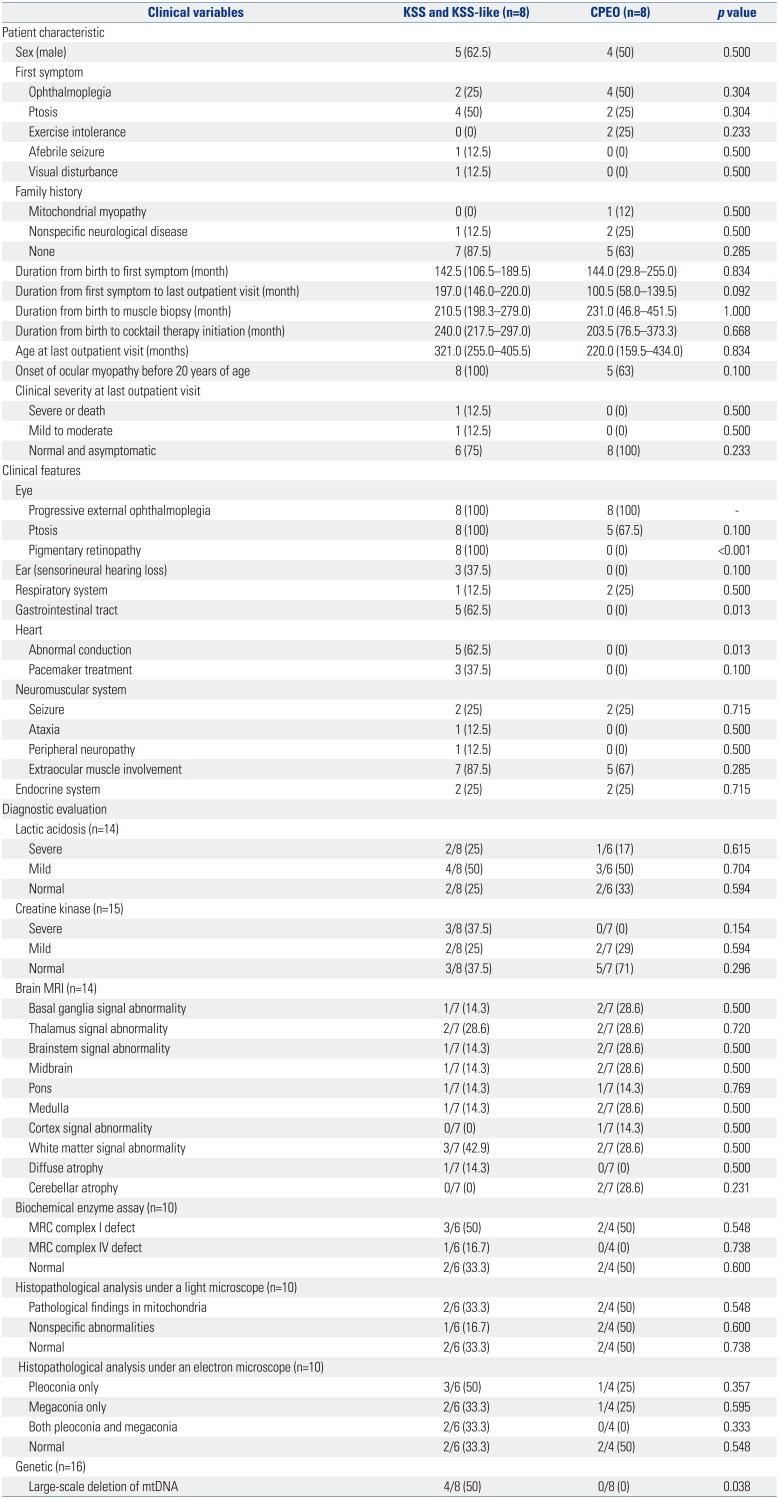
Among the 16 included patients, nine (56.3%) were male (Table 1). The nature of the initial symptoms varied, with ophthalmoplegia (six patients, 38%) and ptosis (six patients, 38%) being the most prevalent, followed by exercise intolerance (two patients, 13%), afebrile seizure (one patient, 6%), and visual disturbance (one patient, 6%). Most patients (12 patients, 75%) had no family history of the disease. The age at onset of the first symptom was 11.9 (6.0–16.4) years. The duration from the first symptom to the last outpatient clinic visit was 11.8 (6.3–17.9) years. Most patients showed ocular myopathy before the age of 20 years (13 patients, 81%). At the last outpatient clinic visit, one patient was deceased, one patient showed moderate clinical severity (wheelchair-bound and/or partially dependent for daily activities), and the other patients showed mild clinical severity (14 patients, 88%).
Clinical features
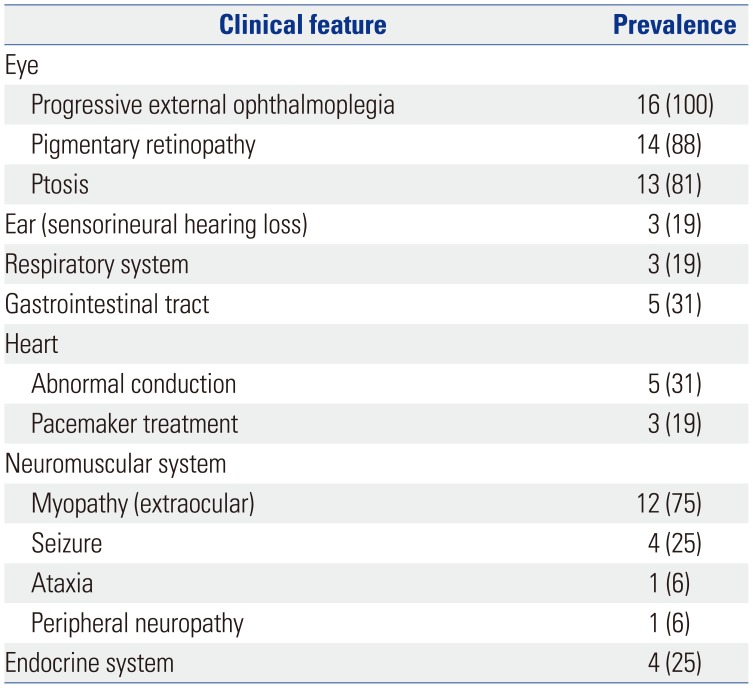
All patients had ophthalmoplegia. Other notable dysfunctions included pigmentary retinopathy or ptosis (14 and 13 patients, respectively), gastrointestinal tract disorders (five patients, 31%), cardiac involvement [five patients (31%) had a cardiac conduction system defect, and three of them had received a pacemaker (19%)], auditory issues (three patients, 19%), and respiratory issues (three patients, 19%) (Table 2). Three quarters of the patients had extraocular myopathy. Endocrine dysfunction was found in four patients (25%) (Table 2).
Diagnostic evaluations
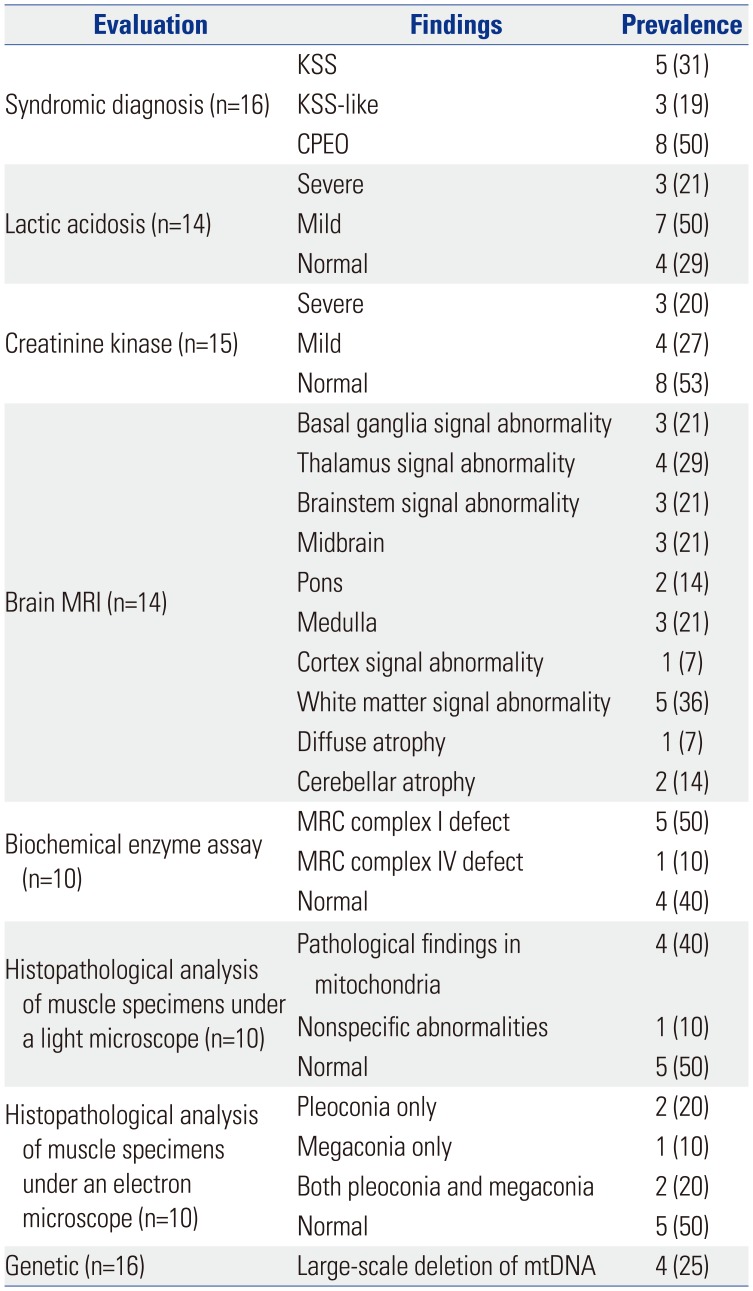
Of the 16 patients with mitochondrial disease and ophthalmoplegia, five (31%) were diagnosed with KSS, three (19%) with KSS-like, and eight (50%) with CPEO (Table 3).
Mild or severe elevations in serum lactate levels were found in 10 patients (71%), while mild or severe elevations in serum creatine kinase levels were observed in seven patients (47%). Brain magnetic resonance imaging (MRI) showed abnormal findings in seven of 14 patients (two did not have MRI results), revealing atrophy or abnormal signals in various areas of the brain.
Muscle biopsy, performed for 10 of the 16 patients, showed abnormal changes under a light microscope in five patients (50%), including findings for nonspecific diseases (one patient) and specific mitochondrial diseases (four patients), such as ragged red fibers and abnormal staining. Abnormal findings under an electron microscope were observed in five patients (50%): these included pleoconia (two patients), megaconia (one patient), and both (two patients). Biochemical evaluation of MRC enzymatic function in the muscle tissue of 10 patients (no results were available for six patients) showed defects in MRC complex I and MRC complex IV in five (50%) and one patient (10%), respectively. Genetic analysis was performed for all patients and revealed large-scale mtDNA deletions in four patients (25%). All four patients carried mtDNA 4977 bp common deletions (nt8482-nt13460).
Comparisons of clinical variables among the three groups
Our study population was divided into three groups (KSS, KSS-like, and CPEO); however, because the group sizes were small, the KSS and KSS-like groups were combined for comparison with the CPEO group (Table 4). There were no significant differences between the KSS/KSS-like group and the CPEO group in terms of sex, type of initial symptoms, family history, and the age at onset of ocular myopathy. In addition, there were no significant differences in the duration from birth to the first symptom, duration from the first symptom to the last outpatient clinic visit, and clinical severity at the last outpatient visit. As opposed to the CPEO group, the KSS/KSS-like group showed pigmentary retinopathy (p<0.001) and cardiac conduction disease (p=0.013). In addition, the KSS/KSS-like group also had gastrointestinal tract disorders, such as abnormal gastrointestinal motility (p=0.013), unlike the CPEO group.
There were no significant differences between the groups with regard to lactic acidosis, elevation of serum creatine kinase levels, brain MRI findings, and results from biochemical enzyme and histopathological assays. The prevalence of large-scale mtDNA deletions was significantly higher in the KSS/KSS-like group than in the CPEO group (p=0.038).
DISCUSSION
The clinical features of mitochondrial diseases vary greatly, creating challenges to their diagnosis and management.10 Peripheral neuropathy, muscle weakness or atrophy, hypotonia or hypertonia, cerebellar ataxia, and leukodystrophy have frequently been described as presenting symptoms.41112 Predominant neuromuscular and ophthalmological symptoms, such as strabismus, ptosis, ophthalmoplegia, optic atrophy with progressive loss of vision, nystagmus, and retinitis pigmentosa, are also observed.11 Mitochondrial diseases with ophthalmoplegia include KSS, CPEO, and Pearson syndrome (PS).13 Because the diagnosis of mitochondrial disease is difficult, there have been few studies about this condition. The most common initial symptom of mitochondrial disease involves the neuromuscular system.411 However, in our study population, the most common initial symptom was an ocular symptom.
In one of the few studies that have reported on this topic, Grönlund, et al.13 found that the prevalence of ophthalmoplegia in patients with mitochondrial disease was 38.6% (22/57), and ophthalmoscopy, which is performed in patients with mitochondrial disease, revealed pigmentation in the macula and/or periphery in 29.6% (16/54) patients. Although a few previous studies have reported on the prevalence of ophthalmoplegia, results vary depending on how the patient groups were formed. Consequently, ophthalmoplegia symptoms may be an important issue in patients with mitochondrial disease. Moreover, it is important to perform an ophthalmological evaluation, including precise retinal examinations, such as ophthalmoscopy, in patients with mitochondrial disease who exhibit ocular symptoms, such as ophthalmoplegia. Six percent of the patients with mitochondrial disease in our study had ophthalmoplegia. In addition, more than 80% patients had pigmentary retinopathy.
The mean age at onset of the first symptom in our study was 12.8 years, which is later than that for other mitochondrial diseases.11 In a study by Han, et al.,14 all patients with Leigh syndrome and esotropia reported disease onset at less than 1 year of age. In another study, Munnich, et al.11 reported that neonatal (before 1 month, 36%) and infantile (1–24 months, 44%) onset were frequently observed. However, most reported cases of KSS have shown that symptoms develop after 10 years of age.151617 These findings indicate that the age of onset of symptoms is a variable clinical characteristic. In mitochondrial diseases, such as KSS and CPEO, where ophthalmoplegia is a major finding, the onset of ocular and other symptoms might be delayed. We, therefore, assume that symptoms and the age of onset vary according to the type of mitochondrial disease and suggest that these factors should be noted and considered in clinical practice.
In a study by García-Cazorla, et al.,18 most of the patients with mitochondrial disease died within the first 3 months (16 of 33). Similarly, the prognosis of other mitochondrial diseases is usually poor.192021 However, the clinical severity of patients in our study was relatively mild. In cases of mitochondrial disease, the prognosis differs depending on the type of syndrome or pattern of organ involvement. The prognosis for mitochondrial diseases with ophthalmoplegia as a main symptom may be better than expected and reported. The reason for this would be that the main symptom is different and its onset age is later. Because of these varying prognoses, care and family counseling should be implemented according to the type of mitochondrial disease. The major prognostic factor in KSS is cardiac involvement, and indeed, complete heart block has been found to be a major cause of death in patients with KSS.22 Eight of the 16 patients in our study underwent cardiac treatment, and three received a pacemaker. Therefore, while the overall prognosis of KSS and CPEO may be good, the prognosis of patients with cardiac involvement could be different. Moreover, there are a few known protocols for determining the prognosis of the various phenotypes and genotypes of this disease, and thus, a protocol for obtaining a disease-specific prognosis is desired.
With regard to the criteria for KSS, our study revealed significant differences in the prevalence of pigmentary retinopathy or cardiac involvement when compared with previous reports.23 The results of our study indicated that the prevalence of gastrointestinal involvement was also significantly different between the CPEO and KSS/KSS-like groups. Gastrointestinal features of mitochondrial disease are expressed as recurrent vomiting, constipation, intestinal pseudoobstruction, and dysphagia.24 Because of these gastrointestinal problems, enteral tube feeding is also frequent in patients with mitochondrial diseases.25 In a paper by Finsterer,26 patients with KSS showed visceral manifestations, such as dysphagia and hepatopathy. KSS and PS may also cause exocrine pancreatic dysfunction and MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes) with intestinal pseudoobstruction. Gastrointestinal manifestations can be primary symptoms of KSS and CPEO, as demonstrated by our results. Therefore, gastrointestinal manifestations may appear as additional symptoms of systemic involvement, which can affect the prognosis. The clinical diagnosis would be improved if the phenotype could be more finely categorized.
Mitochondrial diseases are rare, such that there are limited data about the diseases. Moreover, performing research is difficult. Because mitochondrial disease is heterogenous, it needs better classification and further studies to generalize its homogeneous aspects. In this study, the mean age and follow-up duration were greater than those reported in other studies. In addition, our patients with KSS had gastrointestinal symptoms, which are not part of the existing definition for KSS and may indicate another aspect of systemic involvement. Since our subjects were included in the study based on their phenotypes, not all patients had genotype abnormality. Although large-scale mtDNA deletions were not found in all patients with KSS, it was an objective diagnostic factor for the syndrome. Therefore, a gene study may prove helpful for evaluating patients with KSS. More research on the various types of mitochondrial diseases will increase the understanding of its natural course and improve clinical practice through early diagnosis, patient and family counseling, and selection of appropriate treatments.
 XML Download
XML Download