

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Hepatitis C virus (HCV), an RNA virus belonging to the family of Flaviviridae, threatens the health of over 170 million people worldwide.1 The HCV genome comprises a single positive sense RNA encoding structural protein and nonstructural (NS) proteins, including NS2, NS3, NS4A, NS4B, NS5A, and NS5B, which are essential to RNA replication.2 Despite great advances in therapeutic approaches for treating HCV infection, drug resistance and high cost are still a great challenge.3 Hence, effective strategies that can be widely used for HCV therapy are scarce.
Most viruses, including HCV, express microRNAs (miRNAs), a class of short noncoding RNAs that regulate gene expression and contribute to carcinogenesis.45 Mounting evidence suggests that HCV infection deregulates miRNAs expression, and in turn, miRNAs contribute to HCV processes. miR-122 has been shown to play a noteworthy role in HCV infection, as a promising candidate for anti-HCV treatment, as the inhibition thereof caused a decrease in HCV RNA abundance.67 Apart from miR-122, miR-199a has also been shown to be associated with HCV replication and to be increased in Huh 7.5 cells infected with genotype 2a (JFH1).8 Further, research into cellular miRNA networks has outlined a series of miRNAs, including miR-25 and miR-130, to be functionally related to HCV infection, restricting viral infection and suppressing HCV replication.9 A recent report indicated that miR-373 was beneficial for HCV replication,10 but still more research on the underlying mechanism is needed.
miRNAs are thought to be linked to HCV carcinogenic processes and to impact immune reaction via different signaling pathways.1112 Generally, viral infection can stimulate innate and adaptive immune responses to prevent the host from resisting infection. Faced with viral components, the host responds by activating type 1 interferon (IFN) and pro-inflammatory cytokines to limit viral replication.13 Likewise, HCV infection also leads to the activation of type 1 IFN and dysregulation of a family of IFN-stimulated genes.1415 Although the mechanisms by which HCV infection limits type 1 IFN are largely unknown, recent findings suggest that interferon regulatory factors (IRFs) are a target of HCV1617: IRF5 was found to be down-regulated and to suppress HCV replication.18 Since HCV can provoke immune responses to resist infection, strategies to exploit the host immune response are expected to aid in treating HCV.
In the present study, we demonstrated that the expression of miR-373 is promoted following HCV infection. Functional analysis suggested that addition of miR-373 promoted HCV replication and regulated IRF5 expression, while miR-373 depletion caused the opposite effect. Interestingly, introduction of IRF5 attenuated miR-373-induced replication of HCV. Further mechanistic analysis demonstrated that, in JFH1-infected Huh 7.5 cells, miR-373 depletion lowers HCV replication via activation of type 1 IFN responses by targeting IRF5.
MATERIALS AND METHODS
Specimens
Liver biopsy specimens were obtained from the Department of Transfusion Medicine at Xi'an Central Hospital. Tissues from 8 patients with HCV infection and from 8 patients without related liver diseases were immediately snap-frozen in liquid nitrogen and stored at −80℃ for further study. The forms for tissues collection were approved by the Institutional Research Ethics Committee of Xi'an Central Hospital, and written informed consent was obtained from all patients.
Cell culture and virus
Primary human hepatocytes, human HCV cells Huh 7.5, and 293T cells were purchased from American Tissue Culture Collection (ATCC, Manassas, VA, USA) and were maintained in Dulbecco's Modified Eagle Medium (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Gibco) and 100 U/mL penicillin and streptomycin (Invitrogen, Carlsbad, CA, USA) at 37℃ in a humidified incubator with 5% CO2 during the study.
HCV genotype 2a (JFH1) RNA was prepared by transcription using MEGAscript RNAi kits (Ambion, Austin, TX, USA) and transfected into 4×106 primary human hepatocytes or Huh 7.5 cells using Lipofectamine 2000 (Invitrogen). The cell culture supernatant with JFH1 was filtered through a 0.45-µm filter membrane (Millipore, Billerica, MA, USA) after 96 h infection.
RNA extraction and quantitative reverse transcription PCR
Total RNA from liver tissues or Huh 7.5 cells was extracted using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. The concentration and purity of RNA were determined by a NanaDrop Spectrophotometer (NanoDrop, Wilmington, DE, USA). Subsequently, mRNA or miRNA was used for first strand cDNA synthesis with a Reverse Transcription Kit (Invitrogen) following the manufacturer's instructions. Then, cDNA was diluted and used for quantitative reverse transcription PCR (qRT-PCR) using SYBR Green (Toyobo, Tokyo, Japan) detection with the following amplification protocol: 95℃ for 1 min, 40 cycles of 95℃ for 15 s, and 60℃ for 1 min. Results were analyzed with 2−ΔΔCt method using β-actin and U6 small RNA as housekeeping genes for normalization of HCV mRNA and miR-373. The following primers (Invitrogen) were designed: HCV (Forward, 5′-TCTGCGGAACCGGTGAG TA-3′, Reverse, 5′-TCAGGCACTACCACAAGGC-3′), β-actin (Forward, 5′-AGCAGCATCGCCCCAAAGTT-3′; Reverse, 5′-GGGCACGAAGGCTCATCATT-3′), miR-373 (Forward, 5′-ACACCCCAAAATCGAAGCACTTC-3′; Reverse, 5′-GGAAAGCGCCCCCATTTTGAGT-3′), and U6 (Forward, 5′-GCTTCGGCAGCACATATACTAAAAT-3′; Reverse, 5′-CGCTTCACGAATTTGCGTGTCAT-3′). All assays were repeated in triplicate.
Cell transfection
JFH1-infected Huh 7.5 cells were seeded in six-well plates at a density of 5×104 cells/well and cultured in a humidified incubator with 5% CO2. IRF5 small interfering RNA (si-IRF5) and activation plasmids were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). pcDNA, miR-373 mimics, control mimics (miR-NC), miR-373 inhibitor (anti-miR-373), and control inhibitor (anti-miR-NC) were purchased from Thermo Fisher (Wilmington, DE, USA). The transfection was performed at approximately 80% confluence using Lipofectamine 2000 (Invitrogen), referring to the manufacturer's protocol. Transfection efficiencies were analyzed by qRT-PCR after 24 h.
Luciferase assays
Putative miR-373 targeting IRF5 was predicted by the online software TargetScan (http://www.targetscan.org/vert_72/). The 3′ untranslated regions (3′-UTR) sequences of IRF5 containing the putative binding sites of miR-373 were synthesized by PCR pGL3 luciferase reporter vector (Promega, Madison, WI, USA) to generate the wild-type plasmid (IRF5-wt-3′-UTR). Site-directed mutagenesis of miR-373 complementary bases was cloned into the pGL3-control vector using Q5 Site Directed Mutagenesis Kit (New England Biolabs, Ipswich, MA, USA) to construct mutant-type plasmid (IRF5-mt-3′-UTR). Luciferase assays were performed in 293T cells transfected with IRF5-wt-3′-UTR or IRF5-mt-3′-UTR together with miR-373 mimics or miR-NC using Lipofectamine 2000 according to the manufacturer's protocols. Cells transfected were collected and subjected to luciferase activity analysis using a Luciferase Assay Kit (GeneCopoeia, Rockville, MD, USA) after 48 h.
Western blot
Cell proteins were prepared by RIPA lysis buffer with 1% phenylmethylsulfonyl fluoride and quantified using bicinchoninic acid assay kits (Thermo Fisher) following given instructions. Proteins were denatured at 95℃ for 10 min in protein loading buffer (Invitrogen). Denatured samples were then loaded onto a 10% SDS-PAGE gel and then transferred to polyvinylidene difluoride membranes (Millipore). The membranes were blocked with 5% non-fat milk for 1 h at room temperature and then incubated overnight at 4℃ with primary monoclonal antibodies against NS3, NS5A, IRF5, double-stranded RNA-dependent protein kinase (PKR), 2′–5′-oligoadenylate synthetases (OAS), myxovirus protein A (MxA), or β-actin (Cell Signaling Technology, Danvers, MA, USA). After three washes in Tris-buffer saline containing 0.1% Tween 20 (TBST), membranes were hatched with secondary antibodies conjugated by horseradish peroxidase for 2 h at room temperature, and then washed three times in TBST before incubation with enhanced chemiluminescence chromogenic substrate (GE Healthcare, Amersham, UK) for visualization of immunoreactivity and densitometry analysis using Image Lab software (Bio-Rad, Hercules, CA, USA).
Statistical analysis
All experiments were repeated independently more than three times. Data are presented as means±standard deviations. Statistical analysis was performed using GraphPad Prism 5 (GraphPad Inc., La Jolla, CA, USA) and SPSS 22.0 (IBM Corp., Armonk, NY, USA). Student's t test was used to assess significant differences between groups. Results were considered statistically significant at *p<0.05.
RESULTS
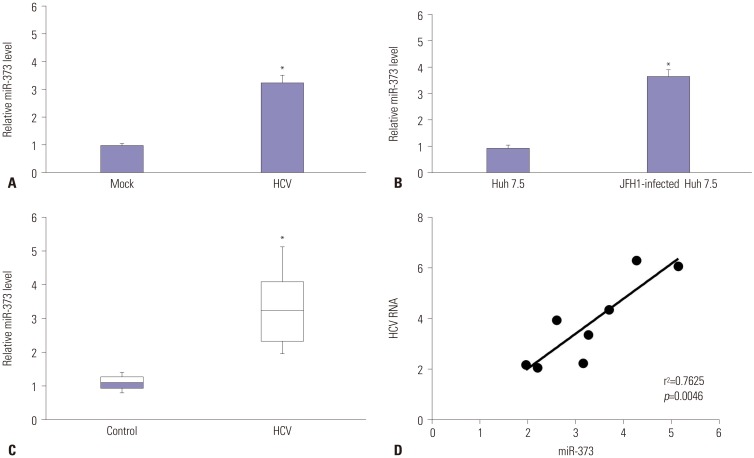
HCV infection enhances miR-373 expression
The major targets and roles of miRNA vary in different pathological conditions. Since human hepatocytes are the natural host of HCV, the abundance of miR-373 was detected between mock-treated and HCV-infected primary human hepatocytes (Fig. 1A). A significant upregulation of miR-373 was exhibited in HCV-infected primary human hepatocytes, compared to mock-treated cells. Similar results were observed for the expression of miR-373 in Huh 7.5 cells, compared between JFH1-infected and mock-treated cells (Fig. 1B). These results suggested that HCV infection promotes the expression of miR-373 in hepatocytes. In addition, HCV infection also resulted in an increased abundance of miR-373 in liver biopsy specimens from patients with HCV infection, compared to non-viral control biopsies (Fig. 1C). To validate the relevance between HCV RNA and miR-373, liver biopsy specimens with HCV infection were introduced repeatedly, revealing a positive correlation between HCV RNA and miR-373 expression levels (r2=0.7625, p=0.0046) (Fig. 1D). Together, these results suggested that HCV infection enhances the abundance of miR-373.
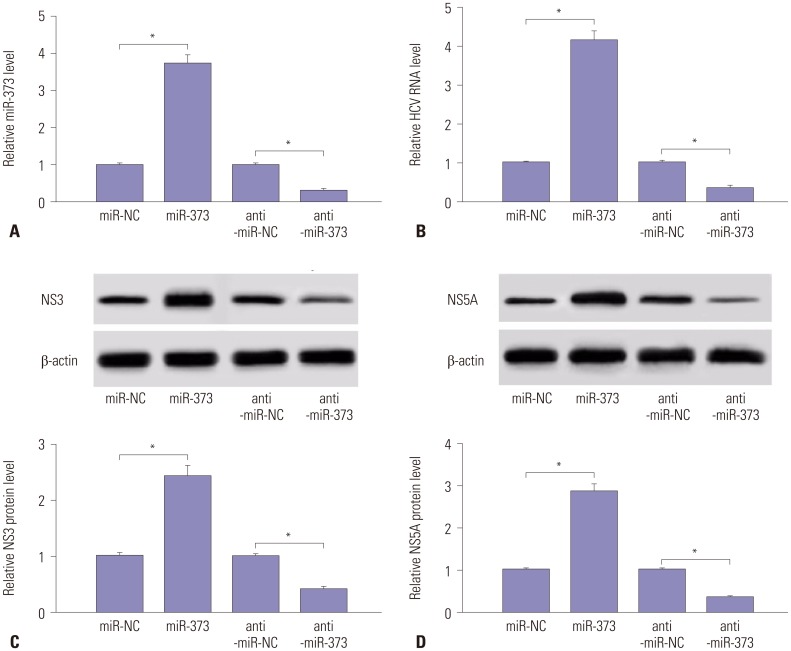
miR-373 facilitates the replication of HCV
To identify whether miR-373 is specially required for the replication of HCV, JFH1-infected Huh 7.5 cells were transfected with miR-373 mimics or inhibitor. The transfection efficiency of miR-373 mimics or inhibitor was confirmed through qRT-PCR assay. Therein, miR-373 was successfully overexpressed by mimics and depleted by inhibitor (Fig. 2A). Furthermore, addition of miR-373 facilitated HCV RNA expression, whereas depletion of miR-373 hindered HCV RNA expression, in JFH1-infected Huh 7.5 cells (Fig. 2B). Further study showed that miR-373 overexpression induced the accumulation of NS3 and NS5A protein in JFH1-infected Huh 7.5 cells, suggesting that miR-373 supports HCV replication and that miR-373 inhibition causes the opposite (Fig. 2C and D). These findings suggested that miR-373 participates in the development of HCV infection.
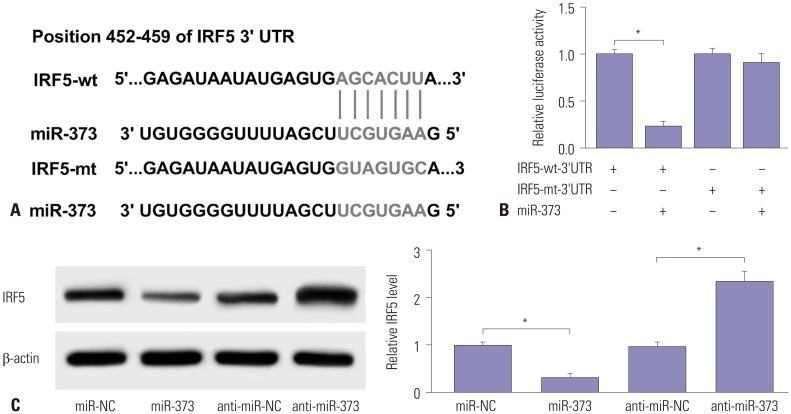
IRF5 is a direct target of miR-373
Having established that miR-373 contributes to HCV replication, we then sought to probe if it interacted with IRF5. Bioinformatics analysis described a putative binding site between miR-373 and the 3′-UTR of IRF5, predicted by the online tool TargetScan, indicating that IRF5 might be a direct target gene of miR-373 (Fig. 3A). Therefore, luciferase assays were used to probe the interaction between IRF5 and miR-373 and presented a remarkable reduction of luciferase activity with the presence of miR-373 mimics in 293T cells transfected with IRF5-wt-3′-UTR, compared to miR-NC. Little effect, however, was observed in those transfected with IRF5-mt-3′-UTR (Fig. 3B). Further, the expression of IRF5 protein was examined by Western blot in JFH1-infected Huh 7.5 cells transfected with miR-373 mimics or inhibitor. As expected, addition of miR-373 limited the expression of IRF5 protein, whereas depletion of miR-373 deregulated IRF5 expression, compared to their counterparts (Fig. 3C). Taken together, these data demonstrated that IRF5 might be a candidate target of miR-373 and that enrichment of miR-373 lowers the expression of IRF5 protein.
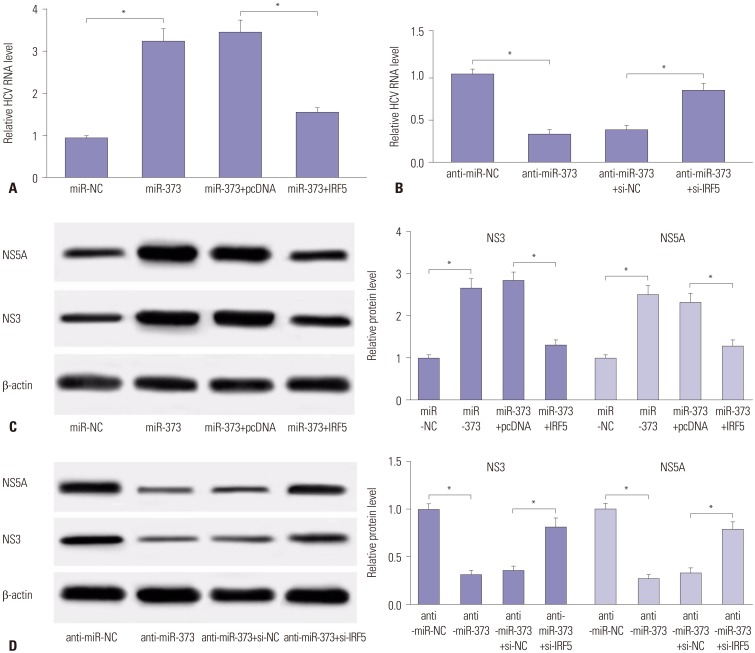
Introduction of IRF5 reverses the positive effects of miR-373 on HCV replication
To further explore whether the positive effects of miR-373 upon HCV replication might be ablated by IRF5, JFH1-infected Huh 7.5 cells were subjected to a short treatment with miR-373 mimics, miR-373+IRF5, miR-373 inhibitor (anti-miR-373), or anti-miR-373+si-IRF5. As a result, the addition of IRF5 led to a striking attenuation of HCV replication in JFH1-infected Huh 7.5 cells, displayed by lower HCV RNA abundances, compared to miR-373 alone (Fig. 4A). On the contrary, depletion of IRF5 reversed the anti-miR-373-mediated inhibition function on HCV replication, causing an increase in HCV RNA expression (Fig. 4B). In addition, the presence of IRF5 elicited a strong decrease in NS3 and NS5A protein abundances in JFH1-infected Huh 7.5 cells transfected with miR-373, compared to the pcDNA treatment group (Fig. 4C), whereas abrogation of IRF5 resulted in the opposite effect (Fig. 4D). These results suggested that depletion of miR-373 impairs HCV replication in the presence of IRF5 in JFH1-infected Huh 7.5 cells.
Inhibition of miR-373 induces activation of type 1 IFN response following restoration of IRF5
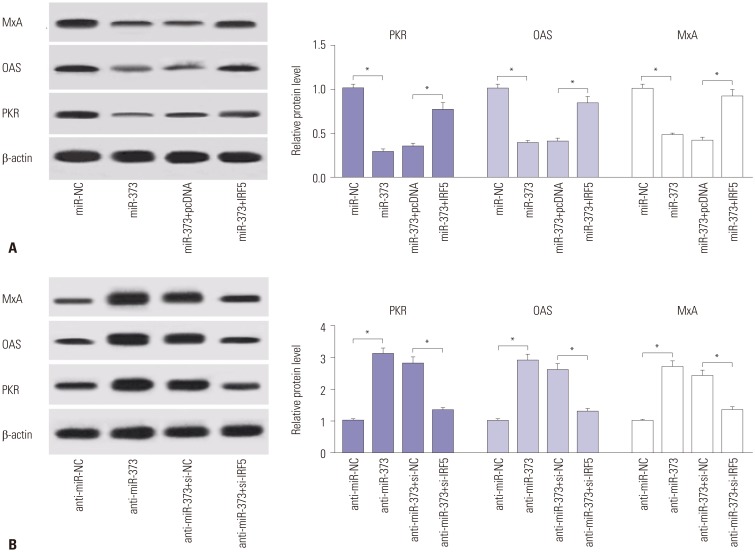
Since IRF5 is known to be associated with type 1 IFN response, the abundances of related proteins (PKR, OAS, and MxA) were detected to investigate the effect of miR-373 and IRF5 on type 1 IFN responses in JFH1-infected Huh 7.5 cells. As a result, accumulation of miR-373 induced marked reductions in PKR, OAS, and MxA protein abundances, while depletion of miR-373 led to the opposite (Fig. 5). Furthermore, the substantial drop in the expression of PKR, OAS, and MxA proteins as a result of miR-373 overexpression was alleviated with the addition of IRF5 (Fig. 5A). Moreover, the promotive effect of miR-373 depletion on type 1 IFN response was abrogated by the introduction of si-IRF5, reflected by a progressive decrease in the abundances of the proteins, compared between anti-miR-373+si-IRF5 and anti-miR-373+si-NC group (Fig. 5B). All findings suggested that miR-373 depletion induces activation of type 1 IFN response via IRF5.
DISCUSSION
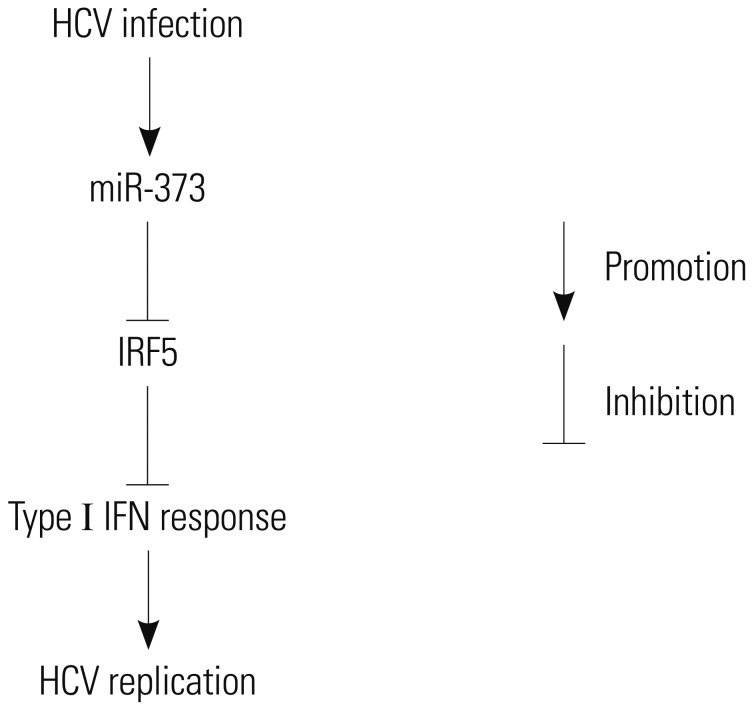
HCV establishes chronic infection in approximately 3% of the global population. There is still a need to explore another driver of HCV infection for the desired antiviral response. In this study, we showed that miR-373 interacts with HCV replication and that this interaction is inhibited by IRF5. Here, we provide the first evidence of HCV replicating poorly in the absence of miR-373 upon activation of the type 1 IFN response via IRF5 (Fig. 6).
According to previous studies, miR-373 is a known requirement for hepatocytes.1920 The discovery that miR-373 is a potential host factor for HCV infection alters the view on how miR-373 interacts with this virus. Here, we showed high expression levels of miR-373 in Huh 7.5 cells infected with JFH1, which is the first HCV strain to produce HCV particles,21 providing an opportunity for HCV progression. miR-373 was reported to facilitate the replication of porcine reproductive and respiratory syndrome virus and herpes simplex virus type 1.2223 Importantly, in this study, the addition of miR-373 promoted HCV replication, revealed by increased abundances of NS3 and NS5A. This is also consistent with findings of other miRNAs that have been found to play an important role in HCV replication, such as miR-122 and miR-155, which were reported as being dysregulated in human HCV cells line or patients.2425 These finding suggest that miR-373 might have a potentially profound impact on the progression of HCV infection. However, more details on the interaction between miR-373 and HCV await discovery.
Generally, functional miRNAs are realized by targeting messenger RNA, causing a decreased abundance of related protein.26 As expected, the existence of binding sites between miR-373 and the 3′-UTR of IRF5 was discovered using TargetScan software. Using another approach to analyze the prediction of IRF5 as a target of miR-373, supported by luciferase assay, we demonstrated that addition of miR-373 hindered the expression of IRF5 protein. Most attention has been paid to IRF5, which was thought to drive inhibition in nasopharyngeal carcinoma and systemic lupus erythematosus.2728 Similarly, during HCV infection, IRF5 could also lower HCV replication in human or mouse hepatocytes.1718 Seeing that IRF5 was also critical for HCV infection, we addressed the role of IRF5 in JFH1-infected Huh 7.5 cells and found that introduction of IRF5 counteracted miR-373-midiated promotion of HCV replication. Despite a large body of results describing the significance of miR-373 and IRF5 on HCV replication, the underlying mechanism was not provided.
Type 1 IFN response is regarded as the first line of host defense and is reflected in the clinically biomarkers PKR29, OAS30, and MxA31, providing an immune microenvironment for HCV32. IRF5 has been shown to be involved in the production of type 1 IFN response in many autoimmune diseases.33 On the other hand, miRNAs have been suggested to be associated with HCV carcinogenic processes and to regulate the immune responses via different signaling pathways.1112 As described by a previous study, depletion of miR-122 contributed to poor IFN therapy response during HCV infection.34 Moreover, miR-155 was also shown to be associated with HCV replication and regulated the immune response via IFN production.35 HCV infection was also shown to boost the expression of miR-146, leading to an alteration of IFN concentrations in sera following HCV infection.36 Similarly, other miRNAs, such as miR-130 and miR-21, have been described as participating in inflammatory and immune responses to HCV infection.37,38 Based on the present findings in this study, we further accessed if miR-373 and IRF5 contribute to activation of type 1 IFN. We found that IRF5 prevented the inhibition of the type 1 IFN response mediated by miR-373 in JFH1-infected Huh 7.5 cells. Recent work has proposed that miR-373 protects against HSV-1 replication via limiting the type 1 IFN response by targeting IFR1.23 Others have suggested that miR-373 knockdown reduces HCV replication via regulating type 1 IFN signaling pathway by targeting Janus kinase 1 and IFR9.10 Contrary with our findings, miR-1225, as an antiviral regulator, was found to enhance type 1 IFN responses by targeting growth factor receptor-bound protein 2-associated binding protein 3.39 Although the findings from this study are interesting, important questions remain about the multiple processes of type 1 IFN response and different genotypes of HCV. Thus, to provide guidance for HCV therapeutically, the related mechanisms await further investigation. Recently, direct-acting antivirals (DAAs) showed effective anti-viral capacity; however, concerns for DAA-resistance mutants are growing.40 Combination with miRNAs might provide additive anti-viral effects, compared with DAAs alone.40
To summarize, this study provides insight into the interaction between miR-373 and HCV infection. Here, we found that HCV infection enhances the expression of miR-373 and that miR-373 depletion limits HCV replication via activation of type 1 IFN response by targeting IRF5 in JFH1-infected Huh 7.5 cells, indicating that miR-373 might present a potential therapeutic targeting opportunity for treating HCV infection.
 XML Download
XML Download