

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Dear Editor,
Congenital prekallikrein (PK) deficiency is a rare autosomal recessive coagulation disorder characterized as either type I or II. Type I is associated with a decrease or absence of PK antigen and activity, while type II is associated with low PK activity and varying antigen levels [1]. PK deficiency presents with prolonged isolated activated partial thromboplastin time (aPTT). Other potential causes of isolated aPTT prolongation without bleeding include presence of lupus anticoagulant (LA) and deficiencies of contact phase factors, including factor XII (FXII) and high molecular-weight kininogen (HMWK) [2]. LA and FXII levels can be easily assessed with laboratory tests, whereas PK and HMWK levels and activity are more challenging to assess. Pre-incubating PK deficient plasma with a surface activator in the aPTT reagent, which causes FXII autoactivation, can bypass the fast PK to kallikrein reaction and kallikrein-mediated FXII activation [3]. During pre-incubation in the aPTT assay, only PK deficiency causes aPTT shortening, a finding that can differentiate PK and HMWK deficiencies. We present the first case, to our knowledge, of congenital PK deficiency in Korea.
This study was approved by the Institutional Review Board of Seoul National University Hospital (No. 1304-085-481), Seoul, Korea. A four-year-old Korean male with tonsil hypertrophy was scheduled to undergo an elective tonsillectomy at Seoul National University Hospital. During the pre-operative evaluation, a marked prolonged aPTT and normal prothrombin time were noted (Table 1). The patient was taking no medication. Personal and family history revealed no bleeding tendency. Mixing test for aPTT showed complete correction, and coagulation factors FVIII, FIX, FXI, and FXII of the intrinsic pathway were all normal. LA was negative. Upon suspicion of PK deficiency, aPTT assays with various pre-incubation times were performed; the aPTT of samples pre-incubated with an aPTT reagent was dramatically shortened (Fig. 1A).
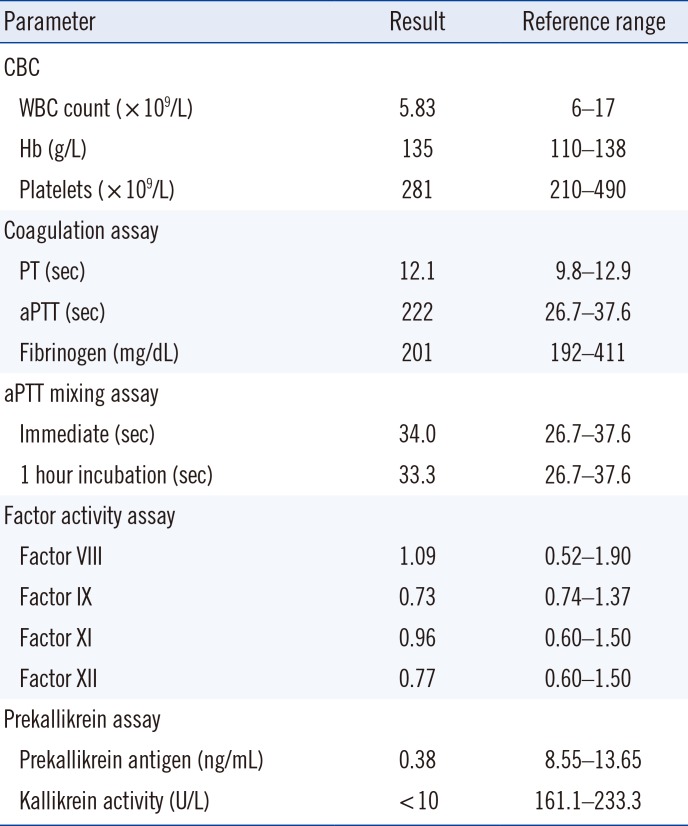
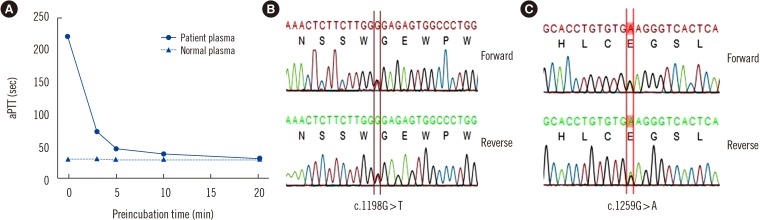 | Fig. 1Results from prekallikrein assays. (A) The patient's and normal plasma were pre-incubated with an activated partial thromboplastin time (aPTT) reagent containing silica activator (SynthASil, Instrumentation Laboratory, Miami, USA) for various times, and clotting times were measured after addition of calcium chloride. Patient plasma pre-incubated with the aPTT reagent showed significant shortening of aPTT. (B) Direct sequencing of KLKB1 showed a c.1198G>T (between red lines) nonsense mutation in exon 11, resulting in premature termination at the 400th amino acid (p.Gly400Ter). (C) Direct sequencing of KLKB1 also showed a c.1259G>A (between red lines) missense mutation in exon 11, leading to the conversion of glycine to glutamine at the 420th amino acid (p.Gly420Glu).
|
Table 1
Patient's laboratory results
![]()
PK antigen level measured by ELISA (SEB801Hu, Cloud-Clone Corporation, Houston, TX, USA) was 0.38 ng/mL, and plasma kallikrein activity measured chromogenically (#K997–100, Biovision, Milpitas, CA, USA) was undetectable. Genomic DNA was extracted from peripheral blood. PCR and direct sequencing of the KLKB1 gene, which encodes prekallikrein, were performed to amplify the coding and flanking regions from exons 2 to 15 [4]. Two variants were detected when comparing the sequence exon with the reference sequence (GenBank Accession Number NM_000892). We detected a novel substitution (c.1198G>T) in exon 11, leading to premature termination of KLKB1 (p.Gly400Ter) (Fig. 1B). This was considered a pathogenic variant according to the 2015 American College of Medical Genetics and Genomics and Association guidelines based on the following factors: PVS1 (predicted null variant), PM2 (absent in population data, Exome Aggregation Consortium, http://exac.broadinstitute.org/), and PP4 (patient's phenotype specific for gene) [5]. A missense mutation (c.1259G>A, p.Gly420Glu) was also found in exon 11 2019.39.2.229(Fig. 1C), which had been detected previously in a family with PK deficiency and caused the loss of PK activity [6].
Functional predictions of the two variants performed using MutationTaster (http://www.mutationtaster.org) revealed them as “disease causing”; therefore, the patient was diagnosed as having type I congenital PK deficiency. He underwent tonsillectomy without any bleeding complication.
Several studies have examined genetic mutations leading to PK deficiency [789]. Mutations in exons 11 and 14, which encode the catalytic domain of the light chain, are frequently observed [7]. In our case, a compound heterozygous mutation was identified in exon 11, as in other cases [789]. Structural analysis showed that the mutants do not contain the catalytic domain of the light chain encoded by exons 11–15. Thus, these mutations may reduce the activity of PK.
Although PK deficiency usually does not increase bleeding tendency, a minor bleeding tendency has been observed [8]. Thrombosis has been reported in fewer than 10 patients with PK deficiency [10]. While FXII deficiency does not cause thrombosis due to defective FXII-induced coagulation activation, PK deficiency may lead to thrombosis, but the mechanism is unclear [78910]. These complications tend to be more commonly reported in type I PK deficiency.
In conclusion, we report a case of congenital PK deficiency with typical results in pre-incubation aPTT assays, caused by a novel pathogenic variant. The possibility of PK deficiency should be considered when treating a patient with isolated aPTT prolongation. The PK deficient patient requires follow-up assessment for the potential complications and should be aware of the risk of unnecessary transfusion.
 XML Download
XML Download