

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Epidemiological typing of pathogenic microorganisms and examining clonal relatedness of pathogens are important components of modern public health infectious disease surveillance [1]. Continued advances in molecular technologies and protocols have revolutionized the practice of epidemiological typing in public health microbiology [1]. Molecular approaches, including multilocus sequence typing (MLST), offer several advantages over traditional typing methods [2].
MLST was designed to overcome the low discriminatory power and poor reproducibility of these methods; it is extensively used because of its superior reproducibility, discriminatory power, and wide applicability of libraries, as well as for inter-laboratory, inter-regional, and inter-national comparisons of pathogenic clones. MLST was first introduced in 1998 by Maiden et al. [3] as a sequence-based counterpart of the traditional multilocus enzyme electrophoresis. Usually, seven MLST loci consisting of approximately 500 bp are indexed, and the unique sequence for each locus is assigned an allelic number as the unit of comparison. The combination of allelic types provides an allelic profile, or sequence type (ST), and is given a numerical designation according to the MLST databases (DBs) [45]. While most other typing procedures involve comparing DNA fragment sizes on gels, MLST sequence data are unambiguous, and the allelic profiles of isolates can easily be compared with those in a large curated central DB via the internet [67].
The major disadvantages of MLST are associated with the high costs and labor-intensity of its data processing [8]. In addition to manual inspection and alignment of sequencing data, the determination of STs according to the web-based DB is also a meticulous process and prone to errors. Large-sized investigational surveillance studies usually involve hundreds to thousands of isolates [91011], and manual assessment of their strain types by MLST can be wearisome. Efforts have been made to simplify the process to prevent clerical errors [1213141516]. However, these systems are costly, or familiarization with their interfaces is difficult. We designed a free automated MLST determination program (MLST typer) based on a Visual Basic for Applications (VBA) macro, which runs on Microsoft Excel (Microsoft Corp, Santa Rosa, CA, USA).
Go to : 
METHODS
Before using MLST typer, the user must ensure that Microsoft Excel 2010–2016 is installed, and that the macro function is enabled
Preparing the allelic type and ST worksheet-format DBs
To query the sequences against the DB in excel worksheets, two types of DBs are constructed: (i) allelic DBs to determine allelic type from the sequence data and (ii) a ST DB to determine STs from the combination of allelic types. Each allelic DB workbook is named after the three-character abbreviation of the species name (e.g., ECO for Escherichia coli and KPN for Klebsiella pneumoniae) and comprises multiple worksheets named after the associated housekeeping genes (i.e., gapA, infB, and mdh). Each worksheet contains two columns: the first for the allelic numbers and the second for their forward-strand reference sequences. In addition, a single ST DB workbook is constructed with multiple worksheets named after each species and the number of housekeeping genes in its MLST scheme adjoined by an underscore (i.e., KPN_7). Each worksheet contains ST numbers in the first column and allelic numbers for the housekeeping genes in the following columns.
Workflow of MLST typer coded via VBA Macro
MLST typer is designed to reflect the traditional workflow of MLST analysis: (i) importing the sequence data into the worksheet, (ii) reverse complement (RC) conversion of reverse sequences, (iii) assembly of forward sequence and RC converted reverse sequence, (iv) determination of allelic types and STs, and (v) summary of the results (Fig. 1).
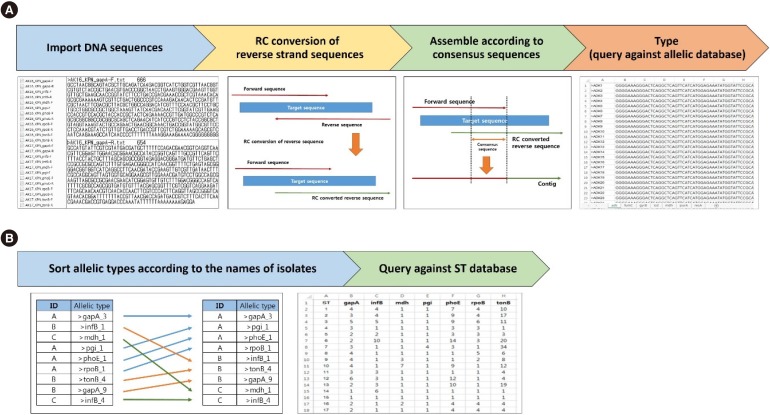 | Fig. 1Schematic process of MLST typer: detailed schematic processes (A) from importing 308 data to allelic typing and (B) from sorting to sequence typing. The software and example files are freely available at: http://medicine.yonsei.ac.kr/en/Research/research_inst/Research_Bacterial/Research_papers/index.asp (article #109) or upon request from the authors at kyunsky@yuhs.ac or parkyj@yuhs.ac.Abbreviations: Contig, contiguous DNA sequence; MLST, multilocus sequence typing; ST, sequence type; RC, reverse-complement.
|
Step 1: Importing the sequence data into the worksheet
FASTA format sequencing data (*.txt) with forward strands and/or reverse strands can be used in this program. Each FASTA file should be formatted with the header line, which begins with the “>” character, isolate name, and/or a unique identifier for the sequence and may also contain additional information, such as length of sequencing product. The following line contains the nucleotide sequences. MLST typer imports the sequencing data from all FASTA format text files located in one designated folder into the worksheet. The file should be named to reflect isolate name, species/gene name, and sequencing strand direction (F-forward or R-reverse).
Step 2: RC conversion of reverse sequences
The reverse sequences should be converted to RC counterparts to query against the prepared allelic DBs with forward-strand reference sequences or for assembly with the forward sequences, and this process is automatically performed by MLST typer. Ambiguity codes other than A, G, C, and T are assumed to be erroneous, and they remain unchanged during the RC conversion processing.
Step 3: Assembly of forward sequence and RC converted reverse sequence
The forward sequence and RC converted reverse sequence are aligned and assembled to form a contiguous DNA sequence (contig) with the user-specified length of consensus sequences. Both ends of the nucleotide sequences can be trimmed in this step. The constructed contigs will be used for MLST determination in the next step. If a consensus sequence region is not found, further steps can be performed with either the forward or RC converted reverse sequence alone, as instructed by the user. For data with only one available sequence, MLST typer will match the sequence against the database.
Steps 4–5: Determination of allelic types and STs and summary of results
The prepared sequences are compared with the reference sequences in the allelic DBs. If a matching sequence is found, an allelic number is assigned. Additionally, MLST typer refers to the ST DB for ST determination of isolates containing complete allelic profiles.
Performance evaluation of MLST typer
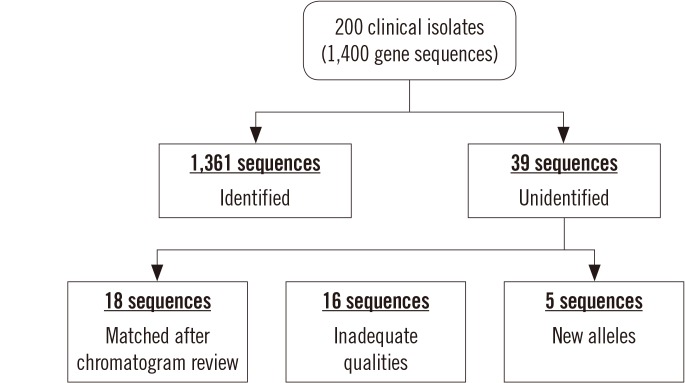
To evaluate the performance of MLST typer, the sequencing data of seven housekeeping genes from 200 clinical isolates were processed: 100 isolates of Escherichia coli, 50 of Klebsiella pneumoniae, and 50 of Staphylococcus aureus. The running time of the sequencing data for 1,400 genes (seven per isolate) was measured. Additionally, the results derived from the MLST typer were compared with those from the manual assessment performed by two expert technicians who routinely perform MLST, and the rates of misidentification or un-identification were calculated. The unidentified sequences were evaluated by reviewing their chromatograms to ensure the adequacy of the sequencing data.
Go to :
RESULTS
Program composition
Main page
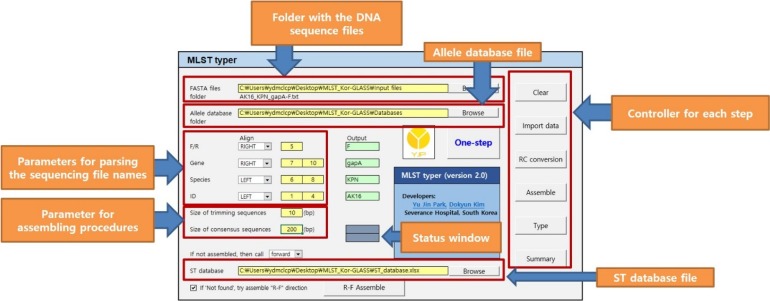
The MLST typer console is located in the Main worksheet (Fig. 2). The browsers for determining folder locations of the sequencing data and allelic DBs are positioned at the top of the console. Parameters for parsing the sequencing file names containing information regarding the forward or reverse direction, gene names, species names, and identifiers are located below. Parsing information can be designated in order from either left or right, and the parsed examples are shown in the mint green sections. The designated species and gene names from the sequencing files should be correlated with the allelic DB names and their worksheets names, respectively. Non-alphabetical/non-numeral values in parsing information are automatically omitted to recognize the intermixed length of the gene names (e.g., mdh and rpoB can be written as mdh_ and rpoB). Size of trimming and consensus sequences need to be established for the assembling procedures. Users may also choose alternative actions when assembly is not possible: to use either the forward strand or RC converted reverse strand for allelic determination or just terminate further processing of that set. The browser for determining the ST DB file is present in the console. The buttons on the right side of the console designate executable processes arranged from top to bottom in order of workflow. The one-step button will perform all processes consecutively. The status window shows which process is being currently handled and what percentage is completed.
Result page
The parameters provided by the user are specified at the top of the Result page, shaded in gray, including the folder locations of sequence files and allelic DBs, the trimming size, and the size of the consensus sequence (Fig. 3A). Color coded columns represent the generated results from each process as follows: blue for I“mport data,” yellow for “RC conversion,” green for “Assemble,” and red for “Type.” The blue columns contain parsed information designated in the console. The imported and RC converted sequences are exhibited in the yellow columns. The “Contig” column in green contains the sequences for further processing; those can be either assembled, forward, or RC converted reverse sequences. The determination results for each gene allelic type are provided in the “Allelic result” column in red, and “Not found” is specified when the processed sequence is not found in the allelic database. The Export button will save a copy in a new Excel workbook.
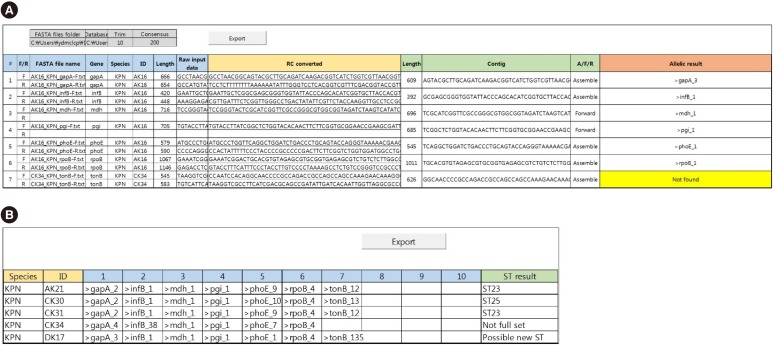 | Fig. 3Example of the Results and Summary worksheet. (A) Example of Results worksheet with assembled sequence and allele typing results. The Result worksheet columns are color-indexed according to each process: importing the DNA sequence (blue), reverse complement (RC) conversion of reverse sequences (yellow), assembling the contig according to consensus sequences (green), and allelic typing (red). (B) Example of Summary worksheet with sorted allelic types and sequence types (ST) results. The Summary worksheet columns are color-indexed according to each process: sort matched allelic types according to isolate names (blue) and find STs queried from the database (green).
|
Summary page
This page aligns allelic types according to their isolate names (Fig. 3B). Species information and isolate name are provided along with the allelic numbers of each housekeeping gene. For ST determination, isolates with complete allelic profiles are queried against the ST DB. If the matching allelic profile is found in the ST DB, a corresponding ST is assigned in the “ST result” column. Alternative messages are specified under the following conditions: “Possible new ST” for a complete allelic profile not matched in the ST DB; “Not full set” for an incomplete allelic profile. The Export button will save a copy in a new Excel workbook.
Miscellaneous
In some cases, the longest contig can be generated by assembling the RC converted reverse sequence before the forward sequence (Fig. 4). Therefore, after the “Type” function has been completed, unidentified sequences can be solved using the “R-F Assemble” function, which creates a contig in reverse-forward-assembly order, followed by the “Type” and “Summary” processes. “R-F Assemble” processed sequences are highlighted in yellow in the “Allelic result” column of the Result page. Users can also decide to include this function in the “One-step” procedure by checking the control box.
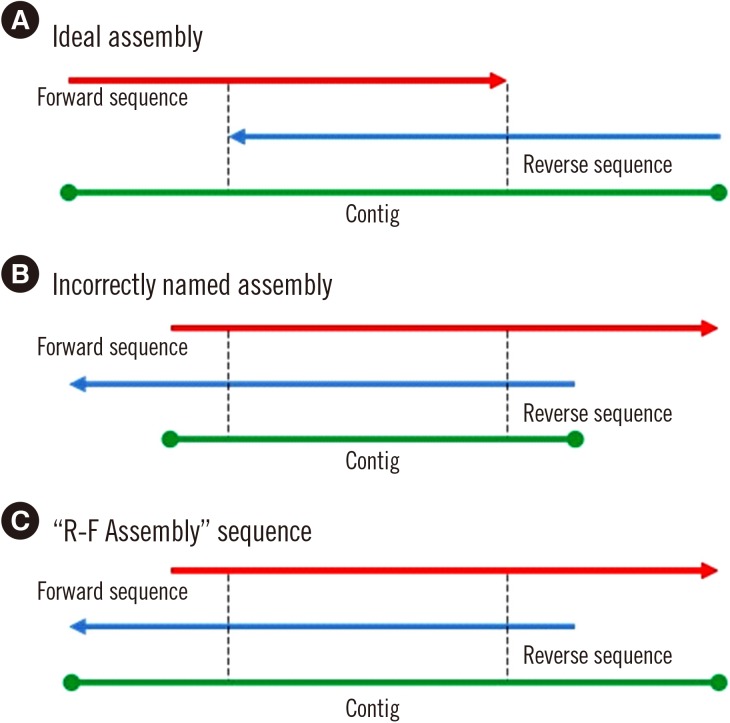 | Fig. 4The “R-F Assembly” process. This is an optional process that can be performed after the main workflow is completed. (A) Ideal assembly provides the longest possible forward-reverse assembled contig. (B) When forward and reverse sequences are incorrectly named, it can result in a shorter than expected contig. (C) The “R-F Assembly” process elongates the assembled contig through reverse-forward order assembly.
|
Performance evaluation of MLST typer
The total running time for 1,400 allelic determinations was 12 min (operated under Microsoft Excel 2013, Windows 7, Intel Core i5 Processor, SSD, 8 GB Memory). Data importing required a substantial amount of time because of the repeated process of opening and closing files, and RC conversion of reverse sequences took most of the remaining running time as the process converts hundreds of individual base characters to their complementary bases. These steps necessitated nearly 10 minutes for processing 1,400 genes. Sequence assembly and determination of the allelic/ST type took only approximately two minutes. The MLST typer showed excellent performance in terms of accuracy in the comparison with manual MLST assessment; the rate of correctly matched allelic types was 97.2% (1,361/1,400; Fig. 5). No misidentified results were observed using the MLST typer.
Go to :
DISCUSSION
We have developed a free automated MLST determination program based on the VBA macro, which runs on Microsoft Excel. Thousands of different programing languages have been created, and many more are being created every year, with popular languages, including Java, Python, PHP, JavaScript, and C#, according to the PopularitY of Programming Language (PYPL) index 2017 (http://pypl.github.io/PYPL.html). Although VBA was ranked in the 12th place, it has several noteworthy advantages, which led us to choose VBA as our programing language. Many technicians prefer working in Microsoft Excel with molecular protocols because of its ease and usefulness in sorting, arranging, filtering, manipulating, and storing data. Furthermore, most people are familiar with the Microsoft Excel platform, and VBA can run in all versions of Microsoft Excel without additional installation or costs.
Writing macros with VBA preempts clerical errors that might increase with many repetitive keystrokes and tasks (Table 1). Additionally, macros reduce the amount of time spent on performing basic computing tasks. MLST typer is able to perform repetitive tasks with simplicity, accuracy, speed, and precision, therefore freeing up microbiologists for more complex problem-solving and idea-generating activities.
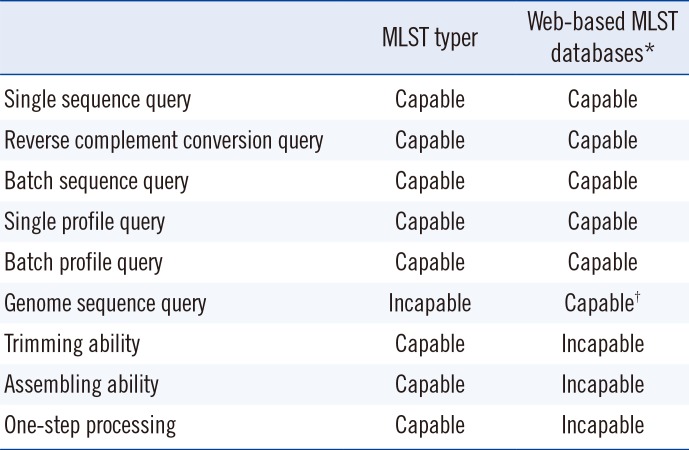
Table 1
Comparison between MLST typer and existing web-based MLST databases
![]()
The flexibility of MLST typer can broaden its scope with minor modifications; it is not solely limited to performing determinations of allelic types and STs. Database files can be altered to fit the needs of many other molecular strain typing protocols, for example, PCR ribotyping of Clostridium difficile isolates and Neisseria gonorrhoeae multi-antigen sequence typing. Additionally, it can be expanded to genotype antimicrobial resistance genes. To enable more practicality, we are planning to provide a web-based MLST typer program in the future, enabling the user to upload FASTA format files and download the processed allelic and ST determination results.
Compared with web-based MLST databases, MLST typer is a fast, off-line standalone program, which requires only Microsoft Excel for execution. Users can manipulate the execution parameters easily using the console, and running all the MLST determination steps is possible by one-step processing. Even though the off-line database used by the MLST typer needs to be created and updated by the user, the time needed to construct a database is compensated by the swift running time of the program.
MLST typer has a few limitations, which may be modified in updated versions. First, there are cases where different genes need different lengths of trimming and consensus sequence parameters. Currently, only one set of assembling parameters is accepted, so the user cannot set different parameters for different genes. Second, inadequate quality of sequencing products may produce erroneous assembly and/or allelic determinations because MLST typer does not have a quality assessment function. For unidentified sequences, quality assessment of sequencing products, such as chromatogram investigation, is advised.
In conclusion, MLST typer can be useful in the domain of epidemiological studies because of its thoroughness in repetitive functions, good compatibility with FASTA type data files, and easy-to-understand outputs for allelic and ST determinations. It has the potential to facilitate large-scale epidemiological studies worldwide.
Go to :
 XML Download
XML Download