

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Thalassemia is a genetic disorder with mutations in the α-globin (chromosome 16) or the β-globin (chromosome 11) gene, and results in a lack of the affected globin chain but accumulation of the unaffected one, which creates globin chain imbalance. Consequently, ineffective erythropoiesis, damage to erythroid membranes, and obstruction of the spleen can occur, accelerating hemolytic anemia or disorders of hematopoiesis [12].
Thalassemia is a serious health problem worldwide, especially in the Mediterranean region, Southeast Asia, and Southern China [134]. In 2008, approximately 20% of the world population carried α+ thalassemia, and 5.2% of the population carried a significant variant of the Hb disorder, including β-thalassemia and α0-thalassemia [1]. Annually, 23,000 cases of β-thalassemia major are diagnosed in newborn infants, and there are 80 million carriers of β-thalassemia worldwide [2]. Approximately 56,000 infants are born annually with severe α- or β-thalassemia, more than half of whom require regular transfusion. Approximately 5,500 annual prenatal deaths are due to hydrops fetalis caused by α-thalassemia major [5].
Because of population migration and transition in recent years, thalassemia patients and carriers are distributed worldwide. The prevalence of thalassemia in Korea was extremely low [6], but the number of immigrants in Korea is increasing. In 2017, there were a total of 2,180,498 immigrants (Chinese: 46.7%, Vietnamese: 7.8%, Thai: 7.0%, and Filipino: 2.7%) in Korea, constituting approximately 4% of the total population [7]. Since thalassemia is a highly prevalent inherited disorder in the Southeast Asian population, its prevalence may increase in Korea in the near future. Hence, epidemiological studies evaluating the prevalence of thalassemia in Korea are necessary, especially since no such studies have been conducted to date.
We investigated the prevalence of thalassemia in Korea in the context of increasing immigration. Our findings could inform guidelines on the diagnosis and management of thalassemia in Korea to improve public health services.
Go to : 
METHODS
Subjects
This was a prospective and observational multicenter study. Between September 2015 and August 2017, we recruited a total of 669 healthy subjects and patients <30 years from seven Korean hospitals: Pusan National University Hospital, Busan; Busan National University Yangsan Hospital, Yangsan; Chonnam National University Medical School, Gwangju; Chonbuk National University Hospital, Junju; Dong-A University Hospital, Busan; Kosin University Gospel Hospital, Busan, and Busan St. Mary Hospital, Busan. Subjects or their parents identified their ethnicity. Subjects were divided into the multiethnic (either or both parents of foreign ethnicity; N=314) or Korean group (both parents of Korean ethnicity; N=355). In the multiethnic group, 80.8% (N=254) had one or more parents who came from Asia. We excluded subjects with ambiguous parental ethnicity, incomplete laboratory test results, or parents known to have hematological malignancies. Subjects' characteristics are described in Table 1. There were no differences in parameters between the two groups.
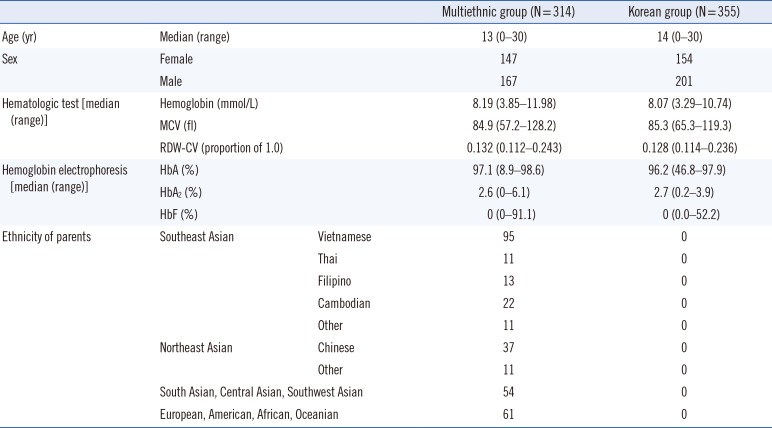
Table 1
Characteristics of the study subjects
![]()
The study protocol was approved by the Institutional Review Board of Pusan National University Hospital (H-1509-001-033). The board decided this study was exempted from the requirement for informed patient or guardian consent when residual serum samples were obtained from subjects during routine testing. The subjects who underwent the genetic testing or their guardians provided written informed consent.
Hematologic testing
Peripheral blood was collected into EDTA-containing tubes, and complete blood count (CBC) was tested simultaneously using an automated cell counter (Sysmex XN-3000; Sysmex, Kobe, Japan). Hb, mean corpuscular volume (MCV), and red blood cell (RBC) distribution width coefficient of variation (RDW-CV) were recorded. An MCV <70 fL and an RDW-CV >14% were classified as abnormal findings. The tests were performed at the recruiting hospital for each subject.
Hb Electrophoresis (EP)
Hb EP was performed on the hemolysate of packed RBCs from blood samples obtained as described above at one core laboratory in Pusan National University Hospital. A complete Hb profile was obtained by capillary zone electrophoresis using CAPIL-LARYS 2 (Sebia, Lisses, France), as per the manufacturer's instructions. Hemolysis was performed by mixing 18 µL of whole blood with 90 µL of alkaline hemolyzing solution (pH 9.4), and the sample was injected by aspiration at the anodic end of the capillary. Hb was separated according to mass:charge ratio under a constant voltage for eight minutes. Hb was detected at 415 nm, resulting in interpretable electropherograms. Electrophoresis quality was tested using the Hb alpha 2 (HbA2) and AFSC commercial control materials (catalog number: PN 4,779) (Sebia). HbA2 >3.5% was considered abnormal. In subjects older than two years, fetal Hb (HbF) >1.2% was considered abnormal, while in those younger than two years, an age-appropriate reference HbF range was used [8].
Genetic testing
Thalassemia was confirmed by genetic testing in subject with low MCV, high RDW-CV, or high HbF and HbA2 levels. Genetic testing was not performed if consent was not obtained or if the amount of sample was insufficient.
Genomic DNA was prepared from whole blood using the Quick-Gene DNA whole blood kit S (Kurabo Industries, Osaka, Japan). The DNA samples were stored at −80℃ until genetic testing. For β-thalassemia, HBB whole gene sequencing was performed. For α-thalassemia, HBA1 and HBA2-specific PCR and sequence analysis, multiple ligation-dependent probe amplification (MLPA), and gap-PCR for deletion analysis were performed. All subjects were tested using MLPA. When MLPA result was −α3.7, −α4.2, --SEA (South-East Asian) type deletion of HBA1 and HBA2 genes, gap-PCR was performed. When the MLPA result was negative, sequence analysis was performed.
MLPA was performed using the P140 HBA -C1 Kit (MRC, Holland, Amsterdam, The Netherlands). Amplified products were analyzed using an ABI 3100 analyzer (Applied Biosystems, Foster City, CA, USA) with GeneMarker software version 1.5.1 (SoftGenetics, State College, PA, USA). Peak heights were normalized, and a deletion or duplication was identified when the normalized peak ratio value was null or two for male subjects.
Sequencing was performed on an ABI 3730 analyzer (Applied Biosystems) using a BigDye Terminator version 3.1 Cycle Sequencing Kit (Thermo Fisher Scientific, Rochester, NY, USA). The sequences were analyzed using SeqScape version 2.5 (Applied Biosystems) and Mutation Surveyor version 3.25 (SoftGenetics). We designed the primer sets using HBA1 (National Center for Biotechnology Information (NCBI) RefSeq: NC_000016.8, NM_000558.3), HBA2 (NC_000016.8, NM_000517.3), and HBB (NC_000011.8, NM_000518.4) DNA sequences. The mutations have been reported according to HbVar: a Database of Human Hemoglobin Variants and Thalassemias [9]. Tests were performed at one core laboratory in Seoul National University Hospital.
Estimation of potential thalassemia carriers in Korea
The number of potential thalassemia carriers in Korea was estimated by multiplying the prevalence of thalassemia in a specific ethnicity by the number of immigrants of that ethnicity. Potential carriers were analyzed for more than 100,100 immigrants categorized by ethnicity. Information on the number of immigrants was extracted from demographic statistics published by the Ministry of Justice [7], and the prevalence of thalassemia in Chinese [4], Vietnamese [10], Thai [11], and American [5] populations was obtained from the literature.
Go to :
RESULTS
In 26 subjects (8.3%) from the multiethnic group and 10 (2.8%) from the Korean group, Hb EP and/or RBC indices showed abnormal findings (Fig. 1). In the multiethnic group, 18 subjects (5.7%) showed low MCV (range: 57.2–69.9 fL) and high RDW-CV (range: 14.0–24.3%), eight subjects (2.5%) showed high HbF (range: 1.6–7.4%), and six (1.9%) showed high HbA2 (range: 3.7–6.1%). In the multiethnic group, two subjects (0.6%) showed high HbF and HbA2, three subjects (1.0%) showed high HbA2 and low MCV, and one subject (0.3%) showed high HbF and HbA2 and low MCV. In the Korean group, two subjects (0.6%) showed low MCV (range: 50.4–65.3 fL) and high RDW-CV (range: 21.6–23.1%), seven subjects (2.0%) showed high HbF (range: 1.3–4.8%), and one subject (0.3%) showed high HbA2 (3.9%). No Korean subject showed two or more abnormal findings.
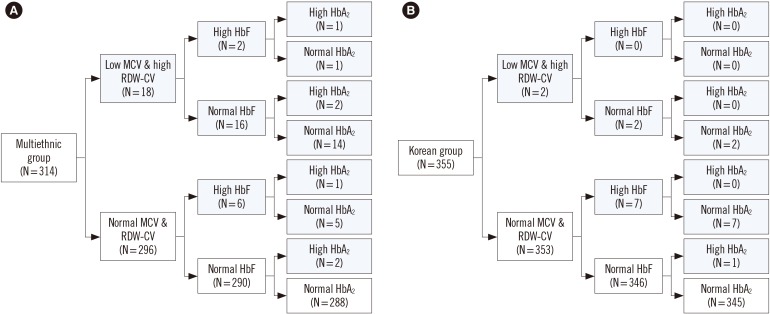 | Fig. 1Hb electrophoresis and RBC indices of the multiethnic group (A) and Korean group (B). Abnormal results are indicated in gray. Abbreviations: HbF, fetal Hb; HbA2, Hb alpha 2; HbA, adult hemoglobin; RBC, red blood cell; MCV, mean corpuscular volume; RDW-CV, RBC distribution width coefficient of variation.
|
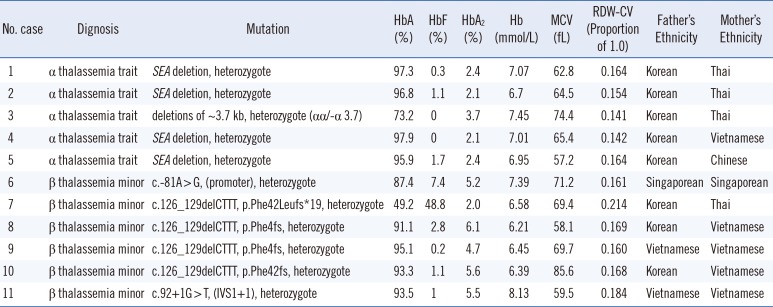
We performed α- and β-globin gene testing in 18 multiethnic subjects and four Korean subjects. Five α-thalassemia carriers (1.5%) were identified, and six subjects (1.9%) were diagnosed as having β-thalassemia minor, all from the multiethnic group. Table 2 shows the subjects' hematological findings and genotypes. The SEA deletion in the HBA1 and HBA2 genes, and c. 126_129delCTTT (p.Phe42fs) mutation of HBB were the dominant inherited mutations (Figs. 2, 3). All six subjects with high HbA2 in the multiethnic group were diagnosed as having thalassemia: one subject with α-thalassemia and five subjects with β-thalassemia minor. Among the 11 subjects with low MCV and high RDW-CV, in the multiethnic and Korean groups, four were diagnosed as having the α-thalassemia trait, and four were diagnosed as having β-thalassemia minor. Among six subjects in the multiethnic and Korean groups with only high HbF and who underwent genetic testing, none were diagnosed as having thalassemia.
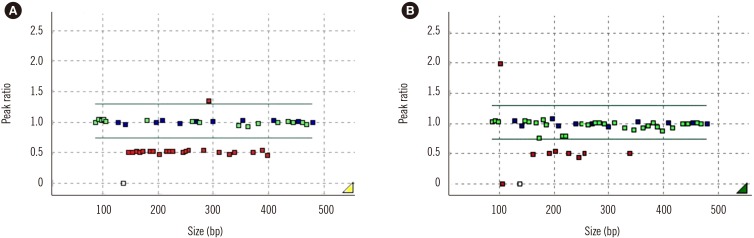 | Fig. 2Multiple ligation-dependent probe amplification results of the HBA1 and HBA2 genes. (A) SEA deletion, heterozygote, (B) Deletions of ~3.7 kb, heterozygote (αα/−α 3.7).Abbreviations: SEA, South East Asian; bp, base pair.
|
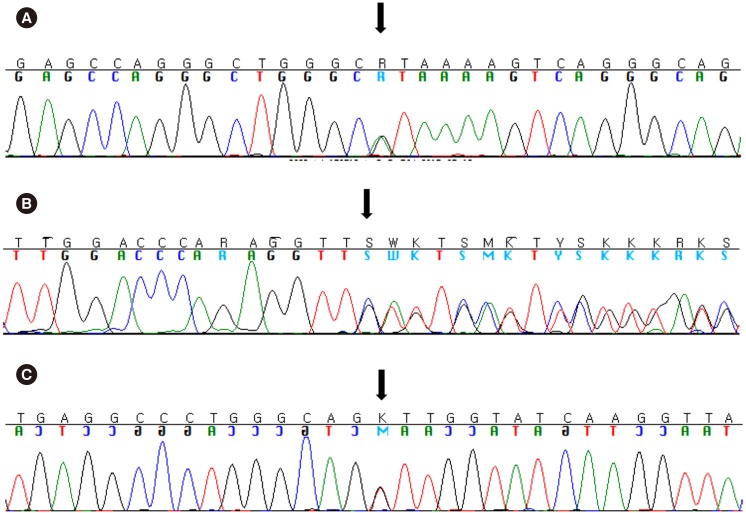 | Fig. 3Direct sequencing results of the HBB gene. (A) c.−81A>G, (promoter), heterozygote, (case 6 in Table 3). (B) c.126_129delCTTT, p.Phe42Leufs*19, heterozygote, (case 7–10 in Table 3). (C) c.92+1G>T, (IVS1+1), heterozygote (case 11 in Table 3).Table 3Estimated number of potential thalassemia carriers in Korea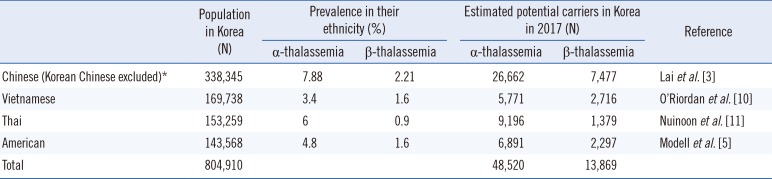
| ||||||||||||||||||||||||||||||||||||||||||||||
Table 2
Hematological findings and results of DNA analysis of α-thalassemia trait and β-thalassemia minor subjects in the study (N=11)
![]()
Among 37 subjects with one or both parents from China, one subject (2.7%) was diagnosed as having β-thalassemia minor. Among 94 subjects with one or both parents from Vietnam, one (1.1%) was diagnosed as having α-thalassemia, and four (4.3%) were diagnosed as having β-thalassemia minor. Among 11 subjects with one or both parents from Thailand, three (27.2%) were diagnosed as having α-thalassemia, and one (9.1%) was diagnosed as having β-thalassemia minor. Among nine subjects with one or both parents from America, none was diagnosed as having thalassemia. As shown in Table 3, the number of potential α-thalassemia and β-thalassemia carriers aomng immigrants was estimated as 48,520 and 13,869, respectively.
Go to :
DISCUSSION
Through genetic testing in individuals with microcytic anemia and abnormal findings on Hb EP in Korea, we found that 1.9% and 1.5% of the multiethnic group were diagnosed as having β-thalassemia minor and α-thalassemia trait, respectively. None from the Korean group was diagnosed as having thalassemia or being a carrier.
Thalassemia results in anemia of various forms, depending on the penetrance of the relevant genetic abnormality [12]. Silent carriers of α-thalassemia are asymptomatic and show an αα/α– genotype, with normal hematologic findings [13]. SEA deletion, the most prevalent mutation in our study, is a large deletion (19.3 kb) that is prevalent in Southeast Asia [1]. It involves the removal of the θ1, α2, and α1 genes [14]. The carrier frequency of α(0)-thalassemia SEA-type deletion has been found to be 3–11% in Thailand depending on the region [15], 2.5–6.1% in China [1], and 13.9% in Japan [16]. In a study on the Korean population, SEA deletion was not reported [17], and only two cases have been reported in patients with South Asian mothers [6]. Since the number of potential thalassemia carriers among immigrants was estimated as 53,740, thalassemia may become a cause of anemia in Korea, unlike before.
Similar to other mutations in α-thalassemia, carriers of the α(0)-thalassemia SEA deletion do not show any clinical symptoms. Gene deletions leading to an αα/-- (cis) (16) or α-/α- (trans) genotype manifest as α-thalassemia minor, characterized by microcytosis without anemia or infrequently with mild microcytic hypochromic anemia [13]. Compound heterozygosity for the α-globin gene with an α-/-- genotype results in HbH disease, characterized by microcytic anemia, hemolysis, and splenomegaly, while an ααT/-- genotype results in HbH disease with normocytic anemia, growth retardation, and splenomegaly. Gene deletions with a --/-- genotype for the α-globin gene lead to α-thalassemia major hydrops fetalis, which is the most severe form of α-thalassemia [13]. Further, approximately 75% of mothers homozygous for the α(0)-thalassemia SEA deletion develop toxemia of pregnancy [18].
The c.126_129delCTTT (p. Phe42fs), the most common mutation of β-thalassemia in our study, is a frameshift mutation that results in a stop codon, terminating translation at the new codon 59. This is a common mutation in China and Southeast Asia, and affects 22–35.3% of the Vietnamese population [1920], 31.7–33.3% of the Chinese population [2122], 37.5% of the Thai population [23], 13.6% of the Japanese population [16], and 4.2% of the Korean population [24]. A β/β0 or β/β+ genotype of the β-globin gene manifests as β-thalassemia minor, leading to microcytosis and anemia. A β0/β+ or a β+/β+ genotype is characteristic of β-thalassemia intermedia, leading to moderate anemia. Cooley's anemia (β-thalassemia major), the most severe form of β-thalassemia, results from a β0/β0, β+/β+, or β+/β0 genotype [13]. It is often called transfusion-dependent thalassemia since patients require repeated blood transfusions throughout their life [12]. In our study, only subjects with β-thalassemia minor and α-thalassemia carriers were identified, not subjects with β-thalassemia major or α-thalassemia. The prevalence of thalassemia major is expected to increase over generations.
Thalassemia is diagnosed on the basis of hematologic findings of microcytic hypochromic anemia and Hb analysis that reveals increased amounts of HbF, HbA2, Hb Barts, and HbH. Identification of pathogenic variants in HBA1, HBA2, and HBB that result in deletion or inactivation of α-globin and β-globin alleles confirms the diagnosis [2526]. However, silent carriers of α-thalassemia and individuals with α-thalassemia minor present with normal hematologic findings or with asymptomatic microcytosis. These conditions are difficult to detect, so we could detect only a few cases of these disorders. Consequently, we observed similar prevalence rates for α-thalassemia and β-thalassemia, unlike in other studies. To assess the true prevalence rate of α-thalassemia carriers, mass screening without hematologic screening is needed. No subject with only high HbF was diagnosed as having thalassemia, and all subjects with high HbA2 were diagnosed as having thalassemia.
Therapies, including allogeneic hematopoietic stem cell transplantation and gene therapy, and pharmacological agents have been developed and tested on patients [2627282930]; however, blood transfusion remains the mainstay of symptomatic management [3132]. Lifelong supportive blood transfusion could lead to iron overload in patients. Thus, close monitoring of serum ferritin levels and iron monitoring by magnetic resonance imaging to prevent iron overload-related organ complications are important [3334]. In Korea, there are no special guidelines for blood transfusion in cases of inherited hemolytic anemia, including thalassemia. Thus, it is important to establish guidelines for blood transfusion and monitoring of iron overload [35].
Children born to parents who are both thalassemia carriers inherit the disease in an autosomal recessive manner and have an increased risk of manifesting thalassemia. Parents and siblings of diagnosed individuals should be screened for carrier status or other mutations, and they should be provided perinatal screening and genetic counseling [133637]. When awareness of thalassemia increases in Korea, timely diagnosis and treatment can be provided.
The prevalence of α-thalassemia was lower than expected. This is probably because we screened blood samples of subjects according to hematologic findings from CBC and Hb EP, and we did not screen for disease carriers. Additional research screening for carriers of α-thalassemia is required.
In summary, we found that 3.4% of the multiethnic group was diagnosed as thalassemia carriers, but none with thalassemia major or severe symptoms. The prevalence of thalassemia may be increasing in young people in Korea because of the increase in the number of immigrants, and the prevalence of thalassemia major is expected to increase over generations. The immigrant population needs to be comprehensively screened as a matter of public health policy to ensure timely diagnosis and treatment of thalassemia. Furthermore, it is imperative that in patients with microcytic hypochromic anemia, which does not respond to iron therapy, a differential diagnosis of thalassemia is evaluated by Hb EP and genetic testing in all Korean patients.
Go to :
 XML Download
XML Download