

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Xanthogranuloma (XG) accounts for 1.6–7% of incidental intracranial lesions discovered on autopsy and XG in the sellar and parasellar region is exceedingly rare [1]. The pathogenesis and exact cause of XG are unknown. Until recently, XG was not included in the WHO classification of central nervous system tumors. XGs are usually small in the sellar region and giant XGs with diameters of greater than 4 cm are extremely rare. The complex anatomy associated with giant XG and the surrounding neurovascular structures can make surgical excision difficult and may increase the risks associated with surgery. There are no accurate reports on the postoperative results, complications, or prognosis of giant XG.
In this paper, we report a case of giant XG that was completely resected without postoperative complications. In addition, we provide evidence supporting the hypothesis that XG originates from Rathke's cleft cyst (RCC). We performed a literature review of all cases of giant XG in the sellar region and described the associated clinical presentations, surgical outcomes, and follow-up results.
CASE REPORT
A 36-year-old man visited our hospital in 2007 due to decreased vision and underwent transphenoidal surgery for pituitary RCC. While receiving treatment at our hospital for hormonal disorders (panhypopituitarism), he presented 2 years after his initial surgery complaining of headache and visual deterioration. At the time of the visit, there were no neurological problems other than a visual field defect and hormonal disorder.
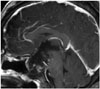
Sellar MRI without contrast revealed a huge, 3.0×4.6×4.6 cm, heterogeneous mass within the sellar region extending up to the third ventricle, right mesial temporal lobe, and retrosellar region (Fig. 1). The suprasellar mass was lobulated with predominantly heterogeneous solid-cystic components on T2-weighted and T1-weighted images. The mass demonstrated relative rim enhancement after gadolinium contrast. There was marked compression and superior displacement of the optic chiasm. There was no evidence of edema in the adjacent parenchyma. A head CT without contrast was obtained and showed a hypodense lesion with wall calcification and erosion of the dorsum sellae (Fig. 1). From the preoperative imaging studies, we suspected craniopharyngioma (CP) and performed surgery. We selected a right orbitozygomatic approach due to the severe optic nerve damage and prominent lateral and retrosellar extension of the tumor. During surgery, we observed severe adhesion of the tumor to the optic nerve, optic chiasm, and the cavernous segment of internal carotid artery with the naked eyes, but the tumor was removed more easily than expected. When the capsule was opened, yellowish-to-brown fluid was found. The pituitary stalk was also well preserved. The tumor is thought to have originated in the sella region.
The postoperative course was uneventful, and the patient's vision slowly improved although it never completely recovered. His preoperative hormonal deficiencies did not improve and continued to be controlled with medications. The postoperative histological findings confirmed to be XG (Fig. 2). Follow-up MRI was performed once a year, and the patient regularly visited our clinic for 6 years after surgery (Fig. 3). Recurrence was not observed during the follow-up period.
DISCUSSION
The most common intracranial site for XG is the choroid plexus, whereas XG is very rare in the sellar and suprasellar regions [2]. We defined giant XG as a lesions with a diameter greater than 4 cm. Including the present case, only 2 cases of giant sellar XG have been reported. Therefore, most preoperative lesions are presumed to be CPs or RCCs.
We consulted Medline PubMed databases to investigate articles related to sellar XG over the past 10 years. Including our patient, we analyzed the clinical features of 29 patients from the papers published in English from 2008 to 2017 [1234567]. The patients included 15 males and 14 females, with a mean age of 28.3 years (range, 5–60 years). Twenty of 29 patients (69%) presented with headache, 15 (52%) had endocrine disturbances, 12 (41%) had visual field disturbances, and one (3%) had diplopia. The lesions were resected via a transsphenoidal approach in 20 patients, and a transcranial approach was used in the other 9 patients. Recurrence occured in only one case over the follow-up periods ranging from 3 to 37 months (mean, 12 months). In most cases, the prognoses were favorable regardless of age, size and location of tumor, surgical approach, and resectability. While the postoperative visual function was improved in our patient, there was no significant change in his hormonal dysfunction.
The origin of sellar XG remains unclear. In 1999, Paulus et al. [8] identified 37 sellar XGs with different histopathologic features and clinical characteristics among 110 patients with adamantinomatous CP. The cholesterol crystals and foreign-body reactions in the cyst walls of XG were histologically similar to radicular cysts, whereas adamantinomatous CP closely resembled odontogenic tumors. XGs and CPs also differed in location, patient age, symptom, and prognosis. Therefore Paulus et al. [8] proposed that XGs represented a distinct entity that is different from both adamantinomatous and papillary CP.
It has also been hypothesized that sellar XG may be represent a secondary reaction caused by the inflammation, hemorrhage, and degeneration of RCC [19]. Ossification has also been associated with secondary granulomatous reactions to RCC [10]. Hama et al. [11] described 20 cases of RCC, 10 of which had inflammatory penetration of the cyst epithelium or subjacent stroma. RCCs were occasionally accompanied by foreign body inflammation around the cyst wall. As a result, sellar XG could be distinguished from adamantinomatous CP, and its characteristics were very similar to those of RCC with inflammatory changes [912]. These findings can be applied to our case, which derived from a secondary progression of RCC. Although the evidence presented above suggest a RCC origin for these lesions, it is not conclusive.
Preoperative differentiation between CP and RCC based on CT and MRI findings is difficult. The cholesterol clefts of a XG may appear hyper-intense in T1-weighted images, although the typical radiological signs of XG remain undefined [1314]. Sugata et al. [15] noted that severe hypopituitarism, hypointensity on T2-weighted MRI imaging, and absence of calcification may also support a preoperative diagnosis of XG. However, the calcification was observed in our case.
Histologically, XG lesions exhibit accumulation of foamy macrophages, multinucleated foreign body giant cells, cholesterol clefts, lymphocytic infiltration, hemosiderin deposits, and fibrous proliferation [8]. Positive immunohistochemical staining for β-catenin is also a reliable marker for differentiation between tumors and cysts of the sellar region. Nuclear β-catenin is found in most adamantinomatous CPs but is absent in papillary CPs and RCCs. Moreover, the ciliated cuboidal and columnar epithelium of RCC indicates that β-catenin immunoreactivity is restricted to the cell membrane [16]. A similar distribution is observed in the fine membranes of arachnoid cysts and cellular elements of XGs [17]. The present case supports the hypothesis that sellar XGs have distinct pathogenic etiologies from CPs, and the two entities should be clearly distinguished (Fig. 2).
In conclusion, our findings indicate that surgical treatment is feasible for any size XG lesion, and the prognosis could be good. In a case of recurrent RCC, XG might be included in differential diagnosis. There is no clear evidence regarding the origin of XG, and so further studies are needed.

 XML Download
XML Download