

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The pathological hallmarks of Alzheimer's disease (AD) include the presence of neuritic plaque, neurofibrillary tangles, and the loss of cortical neurons and synapses [1]. Amyloid beta (Aβ), an amino acid peptide derived from the metabolism of the amyloid precursor protein is a major protein component of the senile plaques in the brains of AD patients. Aβ can promote cellular Ca2+ overload by forming pores in the membrane [2] and inducing membrane-associated oxidative stress [3]. Aβ either directly or indirectly acts on mitochondria to cause increased free radical production and Ca2+ overload. Increasing amounts of evidence implicate changes in [Ca2+]i and oxidative stress as causative factors in Aβ-induced neuronal cell death [4]. Brain zinc accumulation is a prominent feature of advanced AD [5], and increased zinc concentration may serve to accelerate Aβ deposition [6].
Flavonoids have been known to inhibit various ion channels, including Ca2+-permeable ion channels [789]. Cyanidin-3-glucoside (C3G), a component of anthocyanin, belongs to the flavonoid family. C3G has been reported to exert a neuroprotective effect against glutamate-induced neuronal cell death in cultured hippocampal neurons [9] as well as in vitro ischemia-induced neuronal cell death [10], in vivo ischemia-induced neuronal cell death [11], ethanol neurotoxicity in the developing brain [12], and Aβ-induced cell death in cell line [1314]. However, little is known about how C3G affects Aβ-induced neuronal cell death in cultured rat hippocampal neurons.
Aβ25–35 is a short peptide generated from proteolysis of Aβ1–40 that, shows neurotoxic and aggregation effects similar to those of full-length peptides such as Aβ1–40 and Aβ1–42 [15]. The present study aimed to determine whether C3G protects against Aβ25–35-induced neuronal cell death in cultured rat hippocampal cells and pure hippocampal neurons from embryonic day 17 fetal Sprague-Dawley rats using using MTT assay for cell survival and digital imaging methods for Ca2+, Zn2+, MMP, ROS, and caspase-3 assay.
Our results indicate that C3G induced neuroprotection against Aβ25–35-induced apoptosis by reducing multiple signals such as increases of [Ca2+]i and [Zn2+]i, mitochondrial depolarization, the formation of ROS, and the activation of caspase-3 in cultured rat hippocampal neurons.
Go to : 
METHODS
Materials
Dulbecco's modified eagle media, fetal bovine serum, and horse serum were obtained from Gibco-BRL (Grand island, NY, USA). Fura-2 acetoxymethyl ester, dihydroethidine, and rhodamine 123 were purchased from Molecular Probes (Eugene, OR, USA). C3G was prepared from black rice (Oryza sativa L.) by College of Pharmacy, The Catholic University of Korea. All other materials, including Aβ25–35 reagents, were purchased from Sigma (St. Louis, MO, USA).
Culture of primary rat mixed hippocampal cells and pure hippocampal neurons
Isolations of rat hippocampal cells were grown in primary coculture as previously described with minor modifications [9]. Primary cells were obtained from the hippocampi of embryonic day 17 adult maternal Sprague-Dawley rats (250–300 g). All animal research procedures were in accordance with the Laboratory Animals Welfare Act, the Guide for the Care and Use of Laboratory Animals, and the Guidelines and Policies for Rodent Experiment provided by the Institutional Animal Care and Use Committee in the College of Medicine, The Catholic University of Korea (Approval number: CUMC-2015-0171-01). Fetuses were removed on embryonic day 17 from rats that had been anesthetized with urethane (1.3 g/kg body weight, i.p.). Hippocampi were dissected and placed in Ca2+- and Mg2+-free Hank's balanced salt solution adjusted to pH 7.4 with NaOH. Cells were dissociated by trituration through a 5-ml pipette and a flame-narrowed Pasteur pipette.
For digital imaging for Ca2+, Zn2+, MMP and ROS in primary rat mixed hippocampal cells, rat hippocampal cells were grown in primary co-culture as previously described [16] with minor modifications. The cell suspension was centrifuged at 1,000 rpm for 3 min, and the cells were resuspended in Dulbecco's modified eagle media without glutamine, supplemented with 10% fetal bovine serum and penicillin/streptomycin (100 U/ml and 100 µg/ml, respectively). Dissociated cells were plated in six-well culture plates at a density of 50,000 cells/well onto 25-mm-round cover glasses (Fisher Scientific, Pittsburgh, PA, USA) which had been coated with matrigel (0.2 mg/ml) (BD Bioscience) [17]. Neurons and glial cells were grown in a humidified atmosphere of 10% CO2/90% air (pH 7.4) at 37℃. The medium was replaced 72 to 90 h after plating with Dulbecco's modified eagle media supplemented with 10% horse serum and penicillin/streptomycin and fed by an exchange of 25% of the medium after seven days. The cells were cultured without mitotic inhibitors for a minimum of 12 days, and were used between 10 and 13 days in culture for fluorescent dye-based digital imaging methods.
For MTT assay for cell survival and caspase-3 assay in primary rat pure hippocampal neurons, the cells were resuspended in Neurobasal medium (Gibco/Life Technologies, St. Petersburg, FL, USA) supplemented with 2% B27, 1% penicillin/streptomycin (100 U/ml and 100 µg/ml, respectively), 0.5 mM glutamine, and 25 µM glutamate. Dissociated cells were plated at a density of 40,000 cells per well onto 96 wells previously coated with matrigel (0.2 mg/ml). Cells were grown in Neurobasal medium (Gibco/Life Technologies, St. Petersburg, FL, USA) supplemented with 2% B27, 1% penicillin/streptomycin, 0.5 mM glutamine, and 25 µM glutamate at 37℃ in 10% CO2. One-half of the culture medium was changed every three to four days without glutamate [9]. Cells were used between 11 and 13 days in culture.
Calcium imaging
Calcium imaging was performed as described by Yang et al. [9]. The mixed hippocampal cells were loaded with 12 µM fura-2 acetoxymethylester in HEPES-buffered Hank's balanced salt solution containing 0.5% bovine serum albumin for 45 min at 37℃. The HEPES-buffered Hank's balanced salt solution was composed of the following: 20 mM HEPES; 137 mM NaCl; 1.26 mM CaCl2; 0.4 mM MgSO4; 0.5 mM MgCl2; 5 mM KCl; 0.4 mM KH2PO4; 0.6 mM Na2HPO4; 3 mM NaHCO3; and 5.6 mM glucose. Fura-2-loaded cells were alternately excited at 340 and 380 nm. Digital fluorescence images (510 nm) were collected using a computer-controlled, cooled, charge-coupled device camera (1280×1035 binned to 256×207 pixels, Quantix, Photometrics, Tucson, AZ, USA). A ratio of 340/380 nm was calculated from the background-subtracted digital images. Background images were collected at the beginning of each experiment after removing all cells from other areas to the coverslip.
Measurement of Zn2+
In order to monitor the intracellular accumulation of Zn2+, the fluorescent probe FluoZin-3 was used [18]. The mixed hippocampal cells were loaded with 5 µM FluoZin-3 for 45 min, then washed with HEPES-HBSS for 10 min. The fluorescence of FluoZin-3 was detected at 535±25 nm following the excitation of cells at 480±20 nm. Each fluorescence image was detected by the same instruments and methods as those used in calcium imaging. The fluorescence was background-subtracted and normalized to starting values as F/F0.
Measurement of mitochondrial membrane potential and superoxide
In order to measure mitochondrial membrane potential (MMP) and superoxide, the mixed hippocampal cells were loaded with 10 µM rhodamine 123 for 15 min and 10 µM dihydroethidine for 30 min, respectively. Each fluorescence intensity was detected by the same instruments and methods as those used in calcium imaging. While the fluorescence of H2DCFDA was measured following a treatment with amyloid β25–35 or vehicle for 10 min, that of rhodamine was measured following a treatment with amyloid β25–35 or vehicle for 5 min. The fluorescence of rhodamine 123 was measured (excitation 480±10 nm, emission 535±25 nm). The fluorescence of dihydroethidine was measured (excitation 510±40 nm, emission 605±50 nm). The fluorescence was background-subtracted. The formation of ROS, change in MMP, was shown as a percentage of the initial intensity of the fluorescence (F/F0 ×100). The initial intensity of the fluorescence (F0) was measured just prior to the treatment with amyloid β25–35 or vehicle.
Caspase-3 activity assay
After drug treatment, caspase-3 activity assay was carried out according to the manufacturer's protocol (Abcam, ab39401). Pure hippocampal neurons were lysed with 50 µl chilled cell lysis buffer and incubated, centrifuged, and analyzed for total protein using a Bradford assay. The samples were diluted in a ratio of 50 µg protein in 50 µl cell lysis buffer for each assay, then 2× reaction buffer and DEVD-p-NA substrate were added to each sample. After incubation, absorbance was read at a wavelength of 405 nm using a microplate multi-label counter system (Perkin-Elmer, Boston, MA).
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay
Pure hippocampal neuron viability was quantified by measuring the reduction of MTT, which produces a purple formazan product by mitochondrial dehydrogenase in viable cells [9]. Briefly, neurons were treated with amyloid β and/or C3G at 37℃ for 48 h in HEPES-buffered Hank's balanced salt solution. After treatment, fresh HEPES solution containing the MTT at a final concentration of 0.5 mg/ml was added to each well and incubated at 37℃ for 3 h. After removing the medium, dimethyl sulfoxide was used to dissolve the insoluble purple formazan product. Absorbance was measured at a wavelength of 570 nm using a microplate multi-label counter system (PerkinElmer). The absorbance of the formazan that had formed in the non-treated cells in culture medium (control) represented 100% viability.
Statistical analysis
Data are expressed as the mean±S.E.M. Significance was determined using either a Student's t-test or a one-way analysis of variance (ANOVA) followed by a Bonferroni test.
Go to :
RESULTS
Effects of C3G on Aβ25–35-induced cell death in cultured pure rat hippocampal neurons
We first investigated whether C3G affects Aβ25–35-induced neuronal cell death in cultured pure rat hippocampal neurons (Fig. 1A). Pure hippocampal neurons at 11 day in culture were exposed to 20 µM Aβ25–35 containing HEPES-buffered Hank's balanced salt solution for 48 h in either the presence or absence of C3G (5 µg/ml, 10 µg/ml, 15 µg/ml). Treatment with Aβ25–35 (20 µM) for 48 h induced a marked neuronal cell death (p<0.01). Treatment with C3G (5 µg/ml) for 48 h did not significantly affect Aβ25–35-induced neuronal cell death, whereas treatment with higher concentration of C3G (10 µg/ml and 15 µg/ml) significantly increased neuronal cell survival (p<0.05). However, there was no difference seen in neuronal cell survival between 10 µg/ml and 15 µg/ml. Therefore, C3G (10 µg/ml) was used to investigate how C3G induces neuroprotection against Aβ25–35-induced neuronal cell death. Representative phase contrast photomicrographs showed cultured pure rat hippocampal neurons 48 h following co-treatment of C3G (10 µg/ml) with Aβ25–35 (20 µM) at 11 days in culture (Fig. 1B). Treatment with Aβ25–35 (20 µM) for 48 h was found to destroy the cell bodies and processes of hippocampal neurons as compared to those of the vehicle. Co-treatment of C3G (10 µg/ml) with Aβ25–35 was found to inhibit the destruction of the cell bodies and processes of hippocampal neurons. However, treatment for 48 h with C3G (10 µg/ml) alone was not found to affect the hippocampal neurons.
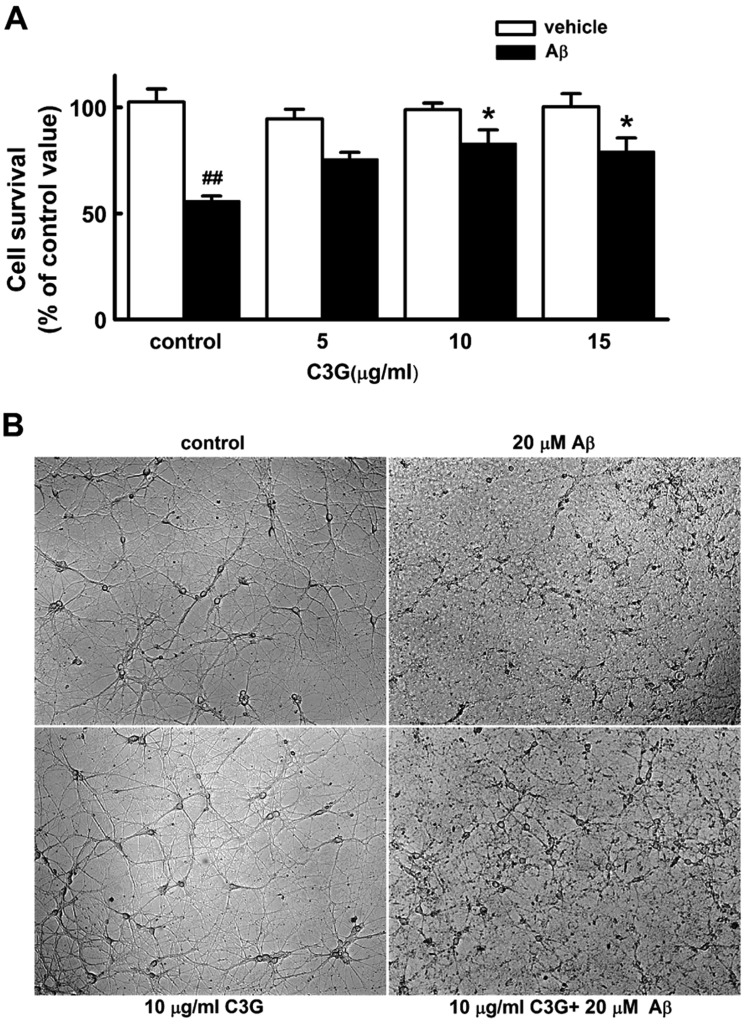 | Fig. 1Effects of C3G on Aβ25–35-induced cell death in cultured pure rat hippocampal neurons.Aβ25–35 (20 µM)-induced neuronal damage was measured through the reduction of MTT in viable cells at 11 days in culture. The bar graph shows the purple formazan product from pure hippocampal neurons after treatment. (A) Effects of C3G on Aβ25–35-induced cell death in the non-treated(control) (vehicle, n=5; Aβ, n=5), 5 µg/ml C3G (vehicle, n=4; Aβ, n=4), 10 µg/ml C3G (vehicle, n=4; Aβ, n=4), and 15 µg/ml C3G (vehicle, n=4; Aβ, n=4)-treated cells in the absence (vehicle) and presence of 20 µM Aβ25–35 for 48 h. (B) Representative phase contrast photomicrographs showing cultured pure rat hippocampal neurons 48 h following co-treatment of C3G with Aβ25–35 at 11 days in culture. Data are expressed as mean±S.E. of experiments. ##p<0.01 relative to control, *p<0.05 relative to control Aβ (ANOVA with Bonferroni test).
|
Effects of C3G on Aβ25–35-induced [Ca2+]i increases
In the following experiment, we investigated how C3G induced neuroprotection against Aβ25–35-induced neuronal cell death. The Aβ25–35-induced [Ca2+]i increase has been proposed as one of the mechanisms involved in Aβ-induced neuronal cell death in cultured cortical neurons [1920] and hippocampal neurons [2122]. Therefore, we determined whether C3G affects the Aβ25–35-induced [Ca2+]i increase in hippocampal neurons. Reproducible [Ca2+]i increases were elicited by applying Aβ25–35 (20 µM) for 5 min at 35 min intervals (Figs. 2A, C). In the previous study, the effects of C3G on ATP-induced Ca2+ signaling reached a maximum at 30 min duration when cells were pretreated with C3G [23]. Therefore, we pretreated neurons with C3G (10 µg/ml) for 30 min in order to determine whether C3G affects Aβ25–35-induced [Ca2+]i increases. Pretreatment with C3G (10 µg/ml) for 30 min was found to significantly inhibit Aβ25–35-induced [Ca2+]i responses (p<0.05) (Figs. 2B, C). These data suggest a possibility that C3G induces neuroprotection against Aβ25–35-induced neuronal cell death by inhibition of [Ca2+]i increases.
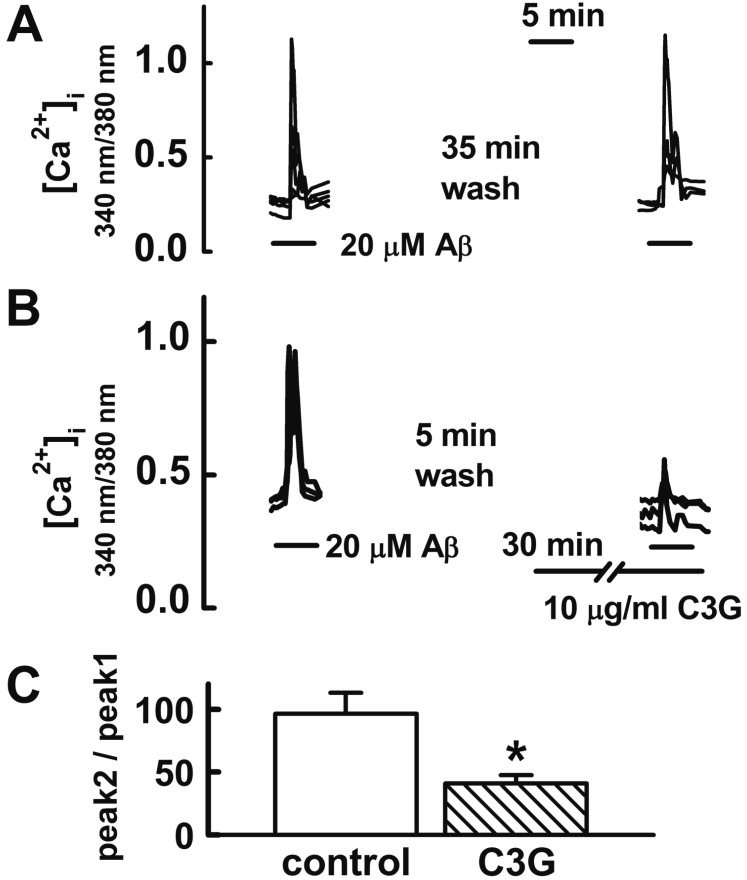 | Fig. 2Effects of C3G on Aβ25–35-induced [Ca2+]i increases.Reproducible [Ca2+]i increases were elicited by applying Aβ25–35 (20 µM) for 5 min at 35 min intervals (A). Pretreatment with C3G (10 µg/ml) for 30 min significantly inhibited Aβ25–35-induced [Ca2+]i responses (B). (C) Summary of the Aβ25–35-induced [Ca2+]i increases in non-treated (control, n=17) and C3G-pretreated (C3G, n=10) cells. Aβ25–35 induced response is presented as a percentage of the initial Aβ25–35 induced response (peak 2/peak 1). Data are expressed as mean±S.E. *p<0.05 relative to control (non-paired Student's t-test).
|
Effects of C3G on Aβ25–35-induced [Zn2+]i increases
Tissue Zn2+ levels are increased in AD, correlating with the levels of brain Aβ [5]. In the present study, C3G inhibited Aβ25–35-induced [Ca2+]i increases. The removal of intracellular Ca2+ with intracellular Ca2+ chelator BAPTA-AM decreases [Zn2+]i and C3G inhibits glutamate-induced [Zn2+]i increases in cultured rat hippocampal neurons by inhibiting [Ca2+]i increases [9]. Therefore, we tested whether C3G inhibits Aβ25–35-induced [Zn2+]i increases (Fig. 3). Treatment with Aβ25–35 (20 µM) for 10 min was found to increase [Zn2+]i increases. Pretreatment with C3G (10 µg/ml) for 30 min was found to significantly inhibit Aβ25–35-induced [Zn2+]i responses (p<0.01).
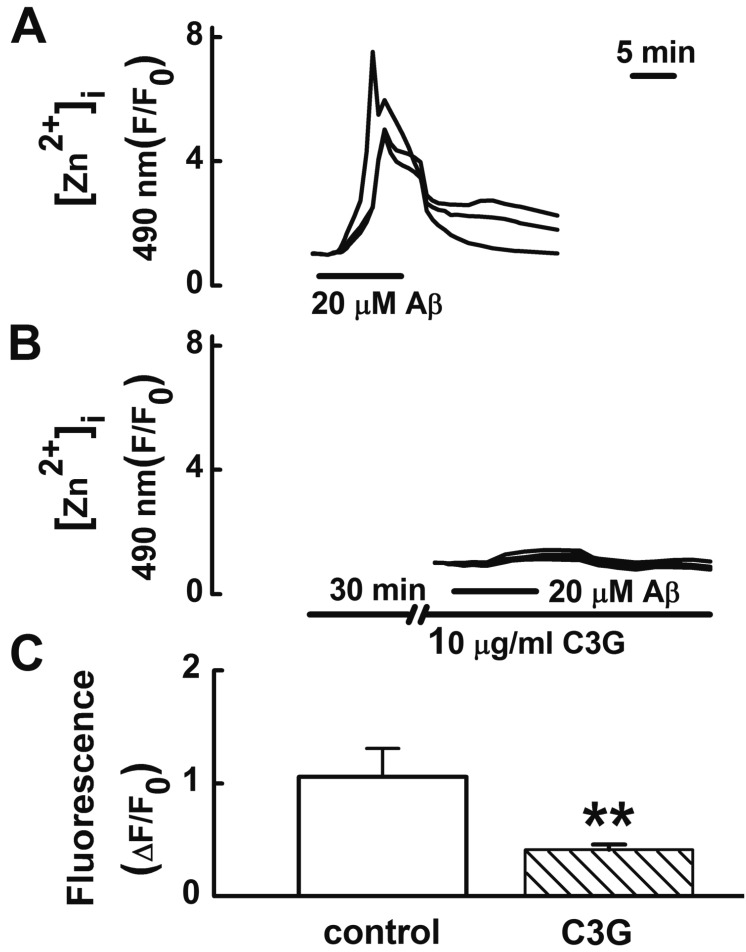 | Fig. 3Effects of C3G on Aβ25–35-induced [Zn2+]i increases.(A) Treatment with Aβ25–35 (20 µM) for 10 min increased [Zn2+]i increases (n=26). (B, C) Pretreatment with C3G (10 µg/ml) for 30 min significantly inhibited the Aβ25–35-induced [Zn2+]i responses (n=22). (C) Aβ25–35 induced response is normalized to the initial value measured before the addition of agonist. Data are expressed as mean±S.E. **p<0.01 relative to control (non-paired Student's t-test).
|
Effects of C3G on Aβ25–35-induced mitochondrial depolarization
An increase in [Ca2+]i [24] or [Zn2+]i [2526] can induce mitochondrial depolarization. Aβ25–35 has been reported to induce an impairment of mitochondrial function [27]. C3G has been reported to inhibit glutamate-induced mitochondrial depolarization as well as [Ca2+]i and [Zn2+]i increases [9]. Therefore, we tested whether C3G inhibits Aβ25–35-induced mitochondrial depolarization (Fig. 4). In this study, treatment with Aβ25–35 (20 µM) for 5 min was found to induce mitochondrial depolarization. Pretreatment with C3G (10 µg/ml) for 30 min was found to significantly inhibit Aβ25–35-induced mitochondrial depolarization (p<0.01) (Fig. 4).
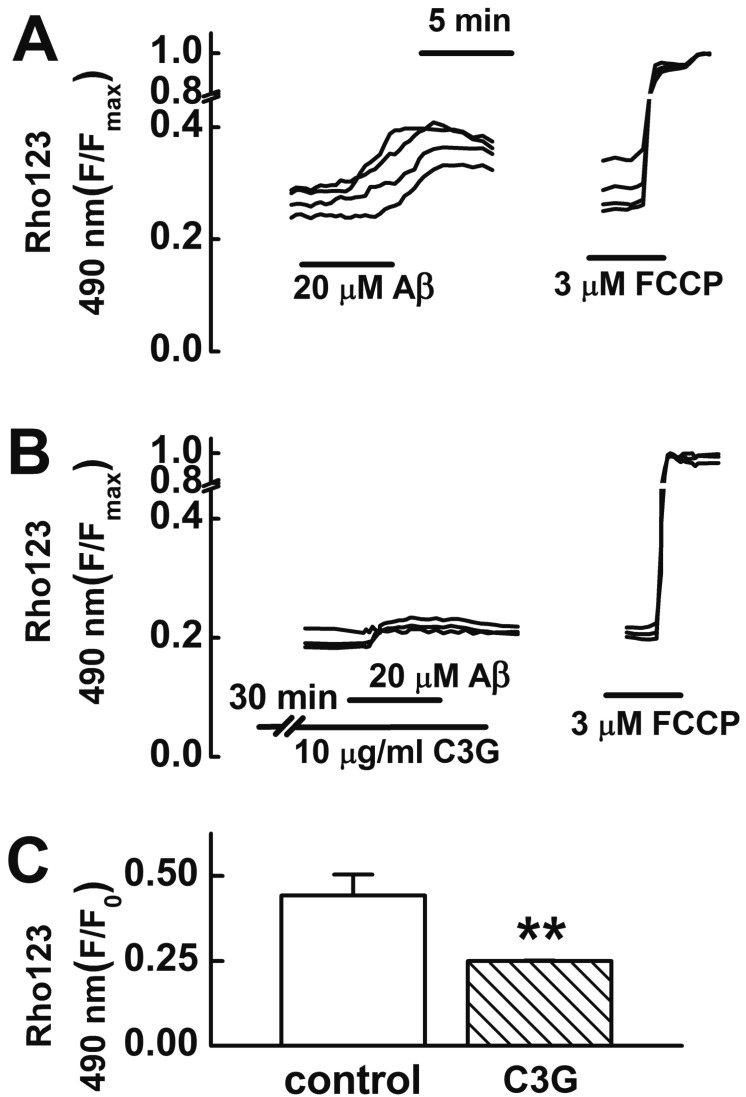 | Fig. 4Effects of C3G on Aβ25–35-induced mitochondrial depolarization.(A) Reproducible depolarization in mitochondrial membrane potential (MMP) was elicited by applying Aβ25–35 (20 µM) for 5 min at 35 min intervals. (B) Pretreatment with C3G (10 µg/ml) for 30 min significantly inhibited Aβ25–35-induced mitochondrial depolarization. (C) Summary of glutamate-induced mitochondrial depolarizations in non-treated (control, n=18) and C3G-pretreated (n=18) cells. Cells were preincubated with 10 µM rhodamine 123 for 15 min. Change in MMP was shown as a percentage of the maximal intensity of rhodamine 123 (3 µM FCCP). Data are expressed as mean±S.E. **p<0.01 relative to control (non-paired Student's t-test).
|
Effects of C3G on Aβ25–35-induced formation of intracelluar ROS
Mitochondrial depolarization induced by [Ca2+]i or [Zn2+]i increase has been reported to lead to the enhanced formation of ROS [24252628]. Aβ25–35 has been reported to induce the formation of ROS [29]. In the present study, C3G inhibited Aβ25–35-induced [Ca2+]i and [Zn2+]i increases, as well as mitochondrial depolarization. Therefore, we tested whether C3G inhibits the Aβ25–35-induced formation of ROS using H2DCFDA (Fig. 5). Treatment with Aβ25–35 (20 µM) for 10 min was found to significantly increase the formation of ROS (p<0.05). Pretreatment with C3G (10 µg/ml) for 30 min was found to block the Aβ25–35-induced formation of ROS (p<0.05).
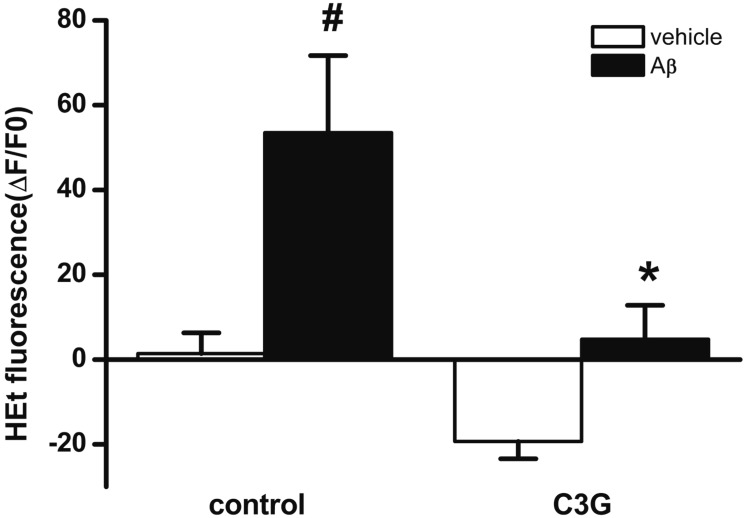 | Fig. 5Effects of C3G on the Aβ25–35-induced formation of intracelluar ROS.Treatment with Aβ25–35 (20 µM) for 10 min significantly increased superoxide formation (vehicle, n=19; Aβ, n=22). Pretreatment with C3G (10 µg/ml) for 30 min blocked Aβ25–35-induced superoxide formation (vehicle, n=16, Aβ, n=14). Superoxide formation was shown as a percentage of the initial intensity of dihydroxyehidine. Data are expressed as mean±S.E. #p<0.05 relative to vehicle (non-paired Student's t-test). *p<0.05 relative to respective control (non-paired Student t-test).
|
Effects of C3G on Aβ25–35-induced activation of caspase-3
Aβ-induced [Ca2+]i increase has been proposed as one of the mechanisms involved in Aβ-induced apoptosis in hippocampal neurons [2130]. In Fig. 1, C3G inhibited Aβ25–35-induced neuronal cell survival. We used a caspase-3 assay in order to determine whether C3G inhibits Aβ25–35-induced neuronal cell death through the activation of caspase-3 (Fig. 6). Treatment with Aβ25–35 (20 µM) for 24 h was found to increase the activation of caspase-3 (p<0.01). Co-treatment with C3G (10 µg/ml) for 24 h was found to significantly inhibit the Aβ25–35-induced activation of caspase-3 (p<0.05).
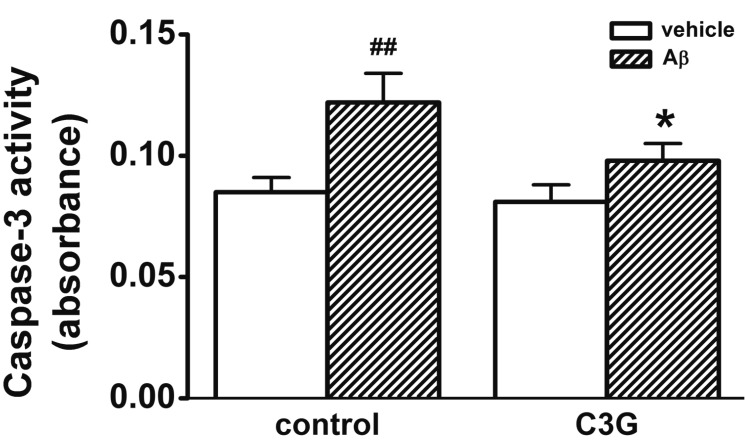 | Fig. 6Effects of C3G on Aβ25–35-induced activation of caspase-3.Treatment with Aβ25–35 (20 µM) for 24 hr increased the activation of caspase-3 (n=6). Co-treatment with C3G (10 µg/ml) for 24 hr significantly inhibited the Aβ25–35-induced activation of caspase-3 (n=4). Data are expressed as mean±S.E. ##p<0.01 relative to vehicle (non-paired Student's t-test) *p<0.05 relative to respective control (non-paired Student's t-test).
|
Go to :
DISCUSSION
This study clearly demonstrated that C3G inhibits Aβ25–35-induced neuronal cell death in cultured rat pure hippocampal neurons. Specifically, C3G was found to inhibit Aβ25–35-induced increases in both [Ca2+]i and [Zn2+]i in cultured rat hippocampal neurons. C3G also inhibited Aβ-induced mitochondrial depolarization and formation of ROS. In addition, C3G inhibited the Aβ-induced activation of caspase-3. These results suggest that C3G inhibits Aβ25–35-induced neuronal cell death by reducing multiple apoptotic signals, such as [Ca2+]i and [Zn2+]i increases, mitochondrial depolarization, the formation of ROS, and the activation of caspase-3.
Aβ has been reported to induce the dysregulation of Ca2+ homeostasis by enhancing two pathways: Ca2+ influx from the extracellular space and Ca2+ release from the intracellular stores. In this study, C3G was found to inhibit Aβ25–35-induced increases in [Ca2+]i and [Zn2+]i in cultured rat hippocampal neurons. Since the removal of intracellular Ca2+ with the intracellular Ca2+ chelator BAPTA-AM inhibited agonist-induced [Zn2+]i increase [9], we assumed that C3G inhibited Aβ25–35-induced [Zn2+]i increase by inhibiting [Ca2+]i increase. Genistein, the active component of soy isoflavones, has been reported to inhibit Aβ25–35-induced [Ca2+]i increases and apoptosis in hippocampal neurons [21]. Recently, Yang et al. [9] reported that C3G inhibits neuronal cell death by inhibiting glutamate-induced [Ca2+]i and [Zn2+]i increases in cultured rat hippocampal neurons. All these data suggest that C3G potentially inhibits Aβ-induced Ca2+ influx through cation-selective channels on the membrane and by modulating the Ca2+-permeable channels, as well as through the release of Ca2+ from the intracellular stores. The inhibitory mechanism of C3G should be investigated in detail in the future.
[Ca2+]i increases induce mitochondrial Ca2+ overload and mitochondrial dysfunction [31]. In the present study, C3G was found to inhibit the Aβ25–35-induced mitochondrial depolarization as well as [Ca2+]i and [Zn2+]i increases. In our previous study, BAPTA-AM or intracellular Zn2+ chelator TPEN inhibited agonist-induced mitochondrial depolarization [9]. These data suggest that C3G inhibits Aβ25–35-induced mitochondrial depolarization by inhibiting [Ca2+]i and [Zn2+]i increases. This result is indirectly supported by previous reports of ours that C3G inhibits glutamate-induced mitochondrial depolarization and neuronal cell death in cultured rat hippocampal neurons [9].
Cytosolic Ca2+ appears to play a pivotal role in the formation of ROS [32]. Mitochondrial Ca2+ uptake induces the formation of ROS [33]. In this study, C3G was found to inhibit the amyloid β25–35-induced formation of ROS, as well as mitochondrial depolarization and [Ca2+]i and [Zn2+]i increases. In a previous study of ours [9], BAPTA-AM or TPEN decreased the agonist-induced formation of ROS. All these data suggest that C3G potentially inhibits the glutamate-induced formation of ROS by inhibiting [Ca2+]i increases. It has been reported that flavonoid genistein inhibits the Aβ-induced formation of ROS [21].
In this study, C3G was found to inhibit the Aβ25–35-induced [Ca2+]i and [Zn2+]i increases, mitochondrial depolarization, and formation of ROS. In addition, C3G was also found to inhibit Aβ25–35-induced caspase-3 activation, as well as neuronal cell death. These results suggest that C3G induces neuroprotection against Aβ25–35-induced apoptosis by reducing multiple signals, such as [Ca2+]i and [Zn2+]i increases, mitochondrial depolarization, the formation of ROS, and the activation of caspase-3. These data are indirectly supported by a report that purple rice extract and its major anthocyanin constituent, cyanidin, induce neuroprotection against Aβ25–35-induced apoptosis in SK-N-SH cells by attenuating the formation of ROS, downregulation of cytochrome C, and activation of caspase [34]. Anthocyanins have also been reported to protect against kainic acid-induced apoptosis in hippocampal neurons via the ROS-activated AMPK pathway, including the activation of caspase-3 [35]. Genistein, one of the flavonoids, has also been reported to ameliorate Aβ25–35-induced apoptosis through various mechanisms, including the inhibition of [Ca2+]i increase, ROS formation, and caspase-3 activation [21].
Aβ25–35 spontaneously inserted into planar lipid membranes forms weakly selective, voltage dependent, ion permeable channels, which act as toxic leaks in plasma and /or intracellular membranes [36]. Aβ generates reactive oxygen species and causes lipid peroxidation and protein oxidation [3738]. These studies suggest that oligomeric Aβ25–35 potentially causes Ca2+ dysregulation, mitochondrial depolarization, disruption of membrane potential, and energy balance via the formation of ion permeable pores and radical-initiated lipid peroxidation, and also may cause neuronal cell death. C3G has been suggested to cross the blood brain barrier and affect neuronal cells [39]. Flavonoids including C3G can have different interaction sites, such as the surface of the membrane and the hydrophobic core of the membrane, based on their chemical properties; this preserves the structure and function of the membrane, including ion channels [4041].
In conclusion, cyanidin-3-glucoside inhibits Aβ25–35-induced [Ca2+]i and [Zn2+]i increases, mitochondrial depolarization, the formation of ROS, and the activation of caspase-3 in cultured rat hippocampal neurons, which are involved in neuroprotection. These results suggest that cyanidin-3-glucoside exerts protective effects against amyloid-induced neuronal cell death by reducing multiple apoptotic signals. The details of how cyanidin-3-glucoside inhibits Aβ25–35-induced signaling pathways at the molecular level should be further investigated in future studies.
Go to :
 XML Download
XML Download