

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Migraine is a prevalent neurological disorder that is characterized by recurrent attacks of severe headache and is accompanied by autonomic symptoms such as photophobia, phonophobia, nausea, and vomiting [12]. Considering that migraine leads to severe disability in the majority of patients, causing considerable burden to their families and society [34], either preventive or abortive medications are needed [5]. The aim of abortive medications like analgesics (non-steroidal anti-inflammatory drugs, acetaminophen), antiemetics (metoclopramide), triptans, or other miscellaneous drugs used against acute migraine attacks is the instantaneous relief of severe headaches and accompanying disorders [67]. Prophylactic medications should be considered for the individual patient, according to the patient factors, such as the frequency of attacks, quality of life, and severity of symptoms, and drug factors, such as the efficacy of the drugs, side effects, and drug interactions [5].
A number of drugs showing the differential mode of actions have been widely used for migraine prophylaxis. These include anticonvulsants, antidepressants, β-adrenoceptor blockers, Ca2+ channel antagonists, and other miscellaneous drugs [8910], suggesting that the several distinct mechanisms are responsible for their prophylactic effects against migraine. Anticonvulsants that block voltage-gated Na+ channels, in particular, valproic acid and topiramate, are proven to be highly effective in migraine prophylaxis [111213]. A clinical study using a cross-over trial compared carbamazepine and placebo and showed that carbamazepine is significantly better than placebo in terms of the responder rate [14]. However, other anticonvulsants that also block voltage-gated Na+ channels, such as lamotrigine and oxcarbazepine, are known to be less effective in migraine prophylaxis [151617], suggesting that the detailed mechanisms underlying the preventive effects mediated by several anticonvulsants are poorly understood.
Carbamazepine is a representative anticonvulsant, but this drug is also widely used for the treatment of chronic pain [18]. In particular, carbamazepine is the first-choice drug for the treatment of trigeminal neuropathic pain, such as nerve injury-induced pain and trigeminal neuralgia [192021]. The generally accepted analgesic mechanism of carbamazepine is an inhibition of voltage-gated Na+ channels, including tetrodotoxin-sensitive and tetrodotoxin-resistant (TTX-R) Na+ channels, resulting in a decreased generation and/or conduction of action potentials in neurons involved in nociceptive transmission [2223]. However, to our knowledge, no study have investigated the direct modulation of TTX-R Na+ channels by carbamazepine in dural afferent neurons, which send their sensory afferents to cranial blood vessels. Therefore, in the present study, we examined the effect of carbamazepine on TTX-R Na+ channels in enzymatically isolated dural afferent neurons identified with the fluorescent dye DiI.
METHODS
Preparation
All experiments complied with the guiding principles for the care and use of animals approved by the Council of Kyungpook National University and the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and every effort was made to minimize both the number of animals used and their suffering.
Sensory neurons of the trigeminal ganglia (TG) innervating the dura were identified after application of the retrograde tracer 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) to the dura, as described previously [24]. Briefly, animals (Sprague Dawley rats, 3–4 weeks old, male) were intraperitoneally anaesthetized with a mixture of ketamine (20 mg/kg) and xylazine (10 mg/kg). The cranial bone overlying the superior sagittal sinus was gently removed by a careful craniotomy using a dental drill, and the dura was exposed. Ten µl of DiI solution (100 mg/ml in DMSO diluted in 1:10 saline) was applied to the dura. One min after the application of DiI solution, a dental resin was placed on the exposed dura to replace the removed cranial bone. The incision was closed with sutures, and penicillin G (100,000 U/kg) and naproxen (10 mg/kg) were intramuscularly injected into the rats to reduce postoperative infection and pain. Seven to 10 days after the DiI application, rats were decapitated under ketamine anesthesia (50 mg/kg, i.p.). The TG was dissected and a pair of ganglia was treated with a standard external solution [in mM; 150 NaCl, 3 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, and 10 Hepes, (pH 7.4 with Tris-base)] containing 0.3% collagenase (type I) and 0.3% trypsin (type I) for 40–60 min at 37℃. Thereafter, TG neurons were dissociated mechanically by triturating with fire-polished Pasteur pipettes in a culture dish (Primaria 3801, Becton Dickinson, Rutherford, NJ, USA). The isolated DiI-positive neurons were used for electrophysiological recordings 1–6 h after preparation (Fig. 1A).

Fig. 1
Effects of carbamazepine on TTX-R Na+ currents.
(A) Typical phase contrast (Ph, left), fluorescent (DiI, middle), and superimposed (right) images of medium-sized DiI-positive neurons. (B) Typical traces of TTX-R Na+ currents in the absence and presence of 100 µM carbamazepine (CBZ). The TTX-R Na+ currents was elicited by electrical stimulation from a VH of −80 mV to −10 mV (100 ms duration) every 5 s (0.2 Hz). Note that carbamazepine more potently decreased the steady-state than peak amplitudes of transient TTX-R Na+ currents. (C) Typical traces of TTX-R persistent Na+ currents in the absence and presence of various concentrations of carbamazepine. The TTX-R persistent Na+ currents were elicited by slow voltage ramp stimulation from a VH of −80 mV to +10 mV (15 mV/s) every 15 s. (D) Concentration-inhibition relationships of carbamazepine for the peak (Ipeak) and steady-state (Isteady) amplitudes of transient TTX-R Na+ currents and slow voltage ramp-induced persistent currents (Iramp). The continuous curve was fitted using a least-squares method. Each point represents the mean and SEM from 10–12 experiments.
![]()
Electrophysiology
All electrophysiological measurements were performed using conventional whole-cell patch recordings and a standard patch-clamp amplifier (Multiclamp 900B; Molecular Devices, Union City, CA, USA). Neurons were voltage clamped at a holding potential (VH) of -80 mV, except where indicated. Patch pipettes were made from borosilicate capillary glass (G-1.5; Narishige, Tokyo, Japan) by use of a pipette puller (P-97; Sutter Instrument Co., Novato, CA, USA). The resistance of the recording pipettes filled with the internal solution was 0.8–1.5 MΩ. Membrane potentials were corrected for the liquid junction potential and the pipette capacitance and series resistance (60–90%) were compensated for. DiI-positive TG neurons were viewed under phase contrast or fluorescence on an inverted microscope (TE2000; Nikon, Tokyo, Japan). Membrane currents were filtered at 2–5 kHz, digitized at 5–20 kHz, except where indicated, and stored on a computer equipped with pCLAMP 10.6 (Molecular Devices). To isolate voltage-gated Na+ currents (TTX-R INa), the internal solution was composed of the following (in mM): 120 CsF, 20 tetraethylammonium (TEA)-Cl, 10 NaCl, 2 EGTA, 2 Mg-ATP, and 10 Hepes (pH 7.2 with Tris-base). The bath solution was composed of the following (in mM): 130 NaCl, 20 TEA-Cl, 2 CaCl2, 1 MgCl2, 10 Hepes, 10 glucose, and 0.01 CdCl2 (pH 7.4 with Tris-base). To record voltage-gated Na+ currents, capacitative and leak currents were subtracted using the P/4 subtraction protocol (pCLAMP 10.6). In current-clamp experiments, both CsF and TEA-Cl were replaced with equimolar KF and KCl, respectively. All experiments were performed at room temperature (22–25℃).
Data analysis
The amplitudes of membrane currents were obtained by subtracting the peak values from the baseline currents using a pCLAMP 10.6 program. The continuous curve for the concentration-inhibition relationship was fitted using a least-squares fit to the following equation: I=1−[Cn/(Cn+IC50n)], where I is the inhibition ratio of TTX-R Na+ currents induced by carbamazepine, C is the concentration of carbamazepine, IC50 is the concentration for the half-effective response, and n is the Hill coefficient. In a subset of experiments, the amplitude of TTX-R Na+ currents, was transformed into conductance (G) using the following equation; G=I/(V−ENa), where ENa is the Na+ equilibrium potential (+40.5 mV) calculated by the Nernst equation. The voltage-activation and voltage-inactivation relationships of TTX-R Na+ channels were fitted to the Boltzmann equations, respectively; G/Gmax=1/{1+exp[(V50,activation−V)/k]} and I/Imax=1−1/{1+exp[(V50,inactivation−V)/k]}, where Gmax and Imax are the maximum conductance and current amplitude, V50,activation and V50,inactivation are half-maximum potentials for activation and fast inactivation, and k is the slope factor. The kinetic data for the recovery from inactivation were fitted to the following equation; I(t)=A0+Afast×[1−exp(−t/τfast)]+Aslow×[1−exp(−t/τslow)], and the kinetic data for the development of inactivation were fitted to the following equation; I(t)=A0+Afast×[exp(−t/τfast)]+Aslow×[exp(−t/τslow)], where I(t) is the amplitude of TTX-R INa at time t , and Afast and Aslow are the amplitude fraction of τfast and τslow, respectively. The weighted time constant (τweighted) was calculated by using the following equation; τweighted=[(τfast×Afast)+(τslow×Aslow)]/(Afast+Aslow). The rheobase (or threshold) currents were determined as the lowest amplitude of the depolarizing current for the generation of action potentials. Numerical values are provided as the mean±SEM using values normalized to the control. Statistical significance of differences in the mean amplitude was tested using Student's paired two-tailed t-test using absolute values rather than normalized ones. Values of p<0.05 were considered significantly different. In order to verify whether the carbamazepine effects were tested in the same conditions, two successive voltage step protocols were applied to the same neurons in the control condition. As shown in Supplementary Fig. S1, TTX-R Na+ currents in response to various types of two successive voltage step protocols were not different statistically.
Drugs
The drugs used in the present study were collagenase, trypsin, tetrodotoxin (TTX), ATP, carbamazepine, and DiI (from Sigma-Aldrich, Seoul, Korea). All solutions containing drugs were applied using the ‘Y–tube system’ for rapid solution exchange [25].
RESULTS
Effects of carbamazepine on TTX-R Na+ channels
The effects of carbamazepine on the peak amplitudes of TTX-R Na+ currents were examined in medium-sized DiI-positive neurons (diameter 30–40 µm, 61.1±10.9 pF, n=152). The transient TTX-R Na+ currents were elicited from a VH of −80 mV by depolarizing step pulses (up to −10 mV, every 5 s). Other voltage-gated ionic currents mediated by TTX-sensitive Na+ channels and voltage-gated Ca2+ channels were blocked by both 300 nM TTX and 100 µM Cd2+, respectively. In these conditions, the application of carbamazepine decreased the peak amplitudes of TTX-R Na+ currents in a concentration-dependent manner. At a 100 µM concentration, carbamazepine decreased the TTX-R Na+ currents slightly but significantly to 96.2±1.0% of the control (n=10, p<0.01, Figs. 1B and D). The half-maximal inhibitory concentration (IC50) of carbamazepine was higher than 1 mM (Fig. 1D). In contrast to the effect on peak amplitudes of TTX-R Na+ currents, carbamazepine effectively decreased the steady-state amplitude of TTX-R Na+ currents in a concentration-dependent manner. At a concentration of 100 µM, carbamazepine substantially decreased TTX-R Na+ currents to 56.7±4.4% of the control (n=10, p<0.01, Figs. 1B and D). The IC50 value for steady-state amplitudes of TTX-R Na+ currents was 165.2±10.5 µM (n=10, Fig. 1D). The steady-state component of TTX-R Na+ currents was completely eliminated in the Na+-free external solution, indicating that it is mediated by TTX-R Na+ channels (data not shown). Additionally, the effects of carbamazepine on persistent Na+ currents was examined, which were elicited by a slow voltage ramp command (−80 mV to +10 mV, 6 s, in every 15 s). Carbamazepine decreased the amplitudes of voltage ramp-induced currents in a concentration-dependent manner (IC50=70.9±6.2 µM, n=12). The voltage ramp-induced persistent Na+ currents were reduced by 100 µM carbamazepine to 36.9±3.0% of the control (n=12, p<0.01, Figs. 1C and D).
Effects of carbamazepine on the voltage-dependence of TTX-R Na+ channels
The voltage-dependence of voltage-gated ion channels is the most fundamental property that determines their activation and inactivation in response to changes in membrane potentials. First, the effects of carbamazepine on the voltage-activation relationship of TTX-R Na+ channels were examined. To evaluate this relationship, the extracellular Na+ concentration was decreased to 20 mM. The TTX-R Na+ currents were elicited by depolarizing test pulses from a VH of −80 mV (10 mV increments, up to +20 mV, every 5 s) in the absence and presence of 100 µM carbamazepine (Figs. 2A and B). After normalizing the conductance of TTX-R Na+ currents to the maximal conductance in the absence of carbamazepine, and then the data were fitted to the Boltzmann function (Fig. 2C). The half-maximal voltage for activation (V50,activation) was not affected by 100 µM carbamazepine. The V50,activation was −25.0±1.6 mV and −24.8±1.5 mV in the absence and presence of carbamazepine, respectively (n=12, p=0.52, Figs. 2Cb and Da). The slope factor k was also not affected by 100 µM carbamazepine (3.8±0.2 mV and 3.9±0.2 mV in the absence and presence of carbamazepine, respectively, n=12, p=0.12, Fig. 2Db), suggesting that the steepness of the voltage-dependence is not affected by carbamazepine.
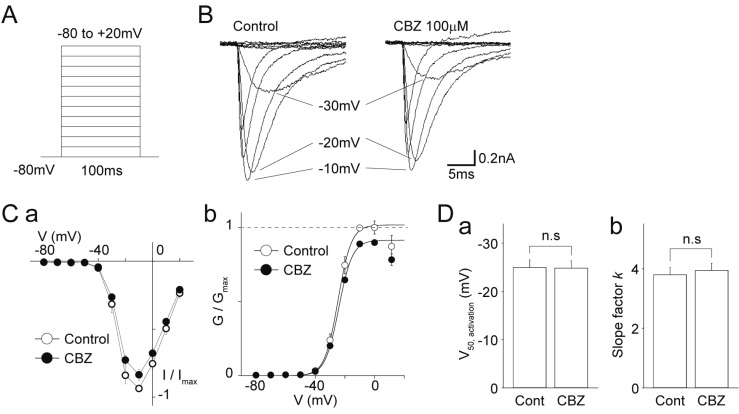
Fig. 2
Effects of carbamazepine on the current-voltage relationship of TTX-R Na+ channels.
(A) A schematic illustration of voltage step pulses. The TTX-R Na+ currents were induced from a VH of −80 mV by 50 ms depolarization pulses from −80 to +20 mV in 10 mV increments. (B) Typical traces of the TTX-R Na+ currents elicited by voltage step pulses in the absence (left) and presence (right) of 100 µM carbamazepine (CBZ). (C) (a) Current-voltage relationships of TTX-R Na+ channels in the absence (open circles) and presence (closed circles) of 100 µM carbamazepine. Each point represents the mean and SEM from 12 experiments. (b) Conductance-voltage relationships of TTX-R Na+ channels in the absence (open circles) and presence (closed circles) of 100 µM carbamazepine. Continuous lines represent the best fit of the Boltzmann function. Each point represents the mean and SEM from 12 experiments. (D) Carbamazepine-induced changes in the half-maximal voltage for activation (V50, activation; a) and the slope factor k (b) of TTX-R Na+ channels. Each column represents the mean and SEM from 12 experiments. n.s, not significant.
![]()
Next, the effects of carbamazepine on the steady-state fast inactivation of TTX-R Na+ channels were examined. The TTX-R Na+ currents were elicited in the absence and presence of 100 µM carbamazepine by depolarizing test pulses (to −10 mV, every 10 s) after 500 ms prepulses from −120 to −20 mV in 10 mV increments (Figs. 3A and B). The peak amplitudes of TTX-R Na+ currents were normalized to the maximal amplitude in the absence of carbamazepine, and these data were fitted to the Boltzmann function (Fig. 3C). The half-maximal voltage for inactivation (V50,inactivation) was shifted towards hyperpolarized potentials by 100 µM carbamazepine. The V50,inactivation was −40.5±2.9 mV and −42.1±2.6 mV in the absence and presence of carbamazepine, respectively (n=10, p<0.01, Figs. 3C and Da). However, the slope factor k was not affected by 100 µM carbamazepine (4.4±0.1 mV and 4.4±0.2 mV in the absence and presence of carbamazepine, respectively, n=10, p=0.90, Fig. 3Db).

Fig. 3
Effects of carbamazepine on the voltage dependence of steady-state fast inactivation of TTX-R Na+ channels.
(A) A schematic illustration of voltage step pulses. The TTX-R Na+ currents were induced by 50 ms depolarization pulses to −10 mV following 500 ms prepulses from −120 to −20 mV in 10 mV increments. (B) Typical traces of the TTX-R Na+ currents elicited by voltage step pulses in the absence (left) and presence (right) of 100 µM carbamazepine (CBZ). (C) Current-voltage relationships of TTX-R Na+ channels in the absence (open circles) and presence (closed circles) of 100 µM carbamazepine. Continuous lines represent the best fit of the Boltzmann function. Each point represents the mean and SEM from 10 experiments. (D) Carbamazepine-induced changes in the half-maximal voltage for inactivation (V50, inactivation; a) and the slope factor k (b) of TTX-R Na+ channels. Each column represents the mean and SEM from 10 experiments. n.s, not significant.
![]()
Furthermore, the effects of carbamazepine on the slow inactivation of TTX-R Na+ channels were examined. The TTX-R Na+ currents were elicited in the absence and presence of 100 µM carbamazepine by depolarizing test pulses (to −10 mV, every 10 s) after 5000 ms prepulses from −100 to −10 mV in 10 mV increments (Figs. 4A and B). The peak amplitudes of TTX-R Na+ currents were normalized to the maximal amplitude in the absence of carbamazepine, and these data were fitted to the Boltzmann function (Fig. 4C). The V50,inactivation was significantly shifted towards hyperpolarized potentials by 100 µM carbamazepine. The V50,inactivation was −43.5±2.1 mV and −47.2±2.2 mV in the absence and presence of carbamazepine, respectively (n=12, p<0.01, Figs. 4C and Da). The slope factor k was also changed by 100 µM carbamazepine (6.5±0.3 mV and 5.7±0.3 mV in the absence and presence of carbamazepine, respectively, n=12, p<0.05, Fig. 4Db).
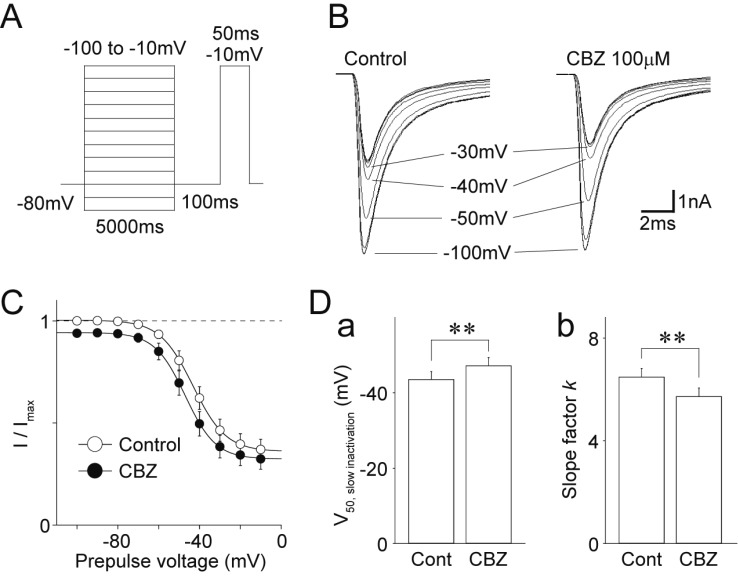
Fig. 4
Effects of carbamazepine on the voltage-dependence of slow inactivation of TTX-R Na+ channels.
(A) A schematic illustration of voltage step pulses. The TTX-R Na+ currents were induced by 50 ms depolarization pulses to −10 mV after 5000 ms prepulses from −100 to −10 mV in 10 mV increments. (B) Typical traces of the TTX-R INa elicited by voltage step pulses in the absence (left) and presence (right) of 100 µM carbamazepine (CBZ). (C) Current-voltage relationships of TTX-R Na+ channels in the absence (open circles) and presence (closed circles) of 100 µM carbamazepine. Continuous lines represent the best fit of the Boltzmann function. Each point represents the mean and SEM from 12 experiments. (D) Carbamazepine-induced changes in the half-maximal voltage for slow inactivation (V50, slow inactivation; a) and the slope factor k (b) of TTX-R Na+ channels. Each column represents the mean and SEM from 12 experiments. **p<0.01.
![]()
Effects of carbamazepine on use-dependent inhibition of TTX-R Na+ channels
The use-dependent inhibition of Na+ channels affects the extent of action potentials generation at higher frequencies. Therefore, the effects of carbamazepine on the use-dependent inhibition of TTX-R Na+ channels were investigated. In these experiments, the TTX-R Na+ currents were elicited by a series of 20 depolarizing pulses (−80 to −10 mV, duration 30 ms) with interpulse intervals of 1000 ms or 200 ms (1 Hz or 5 Hz) between pulses, and the peak amplitudes of TTX-R Na+ currents were normalized to the peak amplitude of the first TTX-R Na+ current (P1) (Figs. 5A and B). The extent of use-dependent inhibition was determined by calculating the amplitude of the 20th TTX-R Na+ currents (P20) relative to P1. As shown in Figs. 5B and C, the use-dependent inhibition of TTX-R Na+ channels expressed in medium-sized DiI-positive neurons was negligible. The P20/P1 ratios in the absence of 100 µM carbamazepine were 0.96±0.03 and 0.94±0.03 after 1 Hz and 5 Hz stimulation, respectively (n=11). Carbamazepine (100 µM) had a minor effect on the use-dependent inhibition of TTX-R Na+ channels; the P20/P1 ratios were slightly decreased to 0.94±0.05 and 0.90±0.04 after 1 Hz and 5 Hz stimulation, respectively (n=11, Figs. 5B and C).

Fig. 5
Effects of carbamazepine on the use-dependent inactivation of TTX-R Na+ channels.
(A) Typical traces of TTX-R Na+ currents elicited by 20 successive voltage step pulses (−80 mV to −10 mV, 30 ms duration, 5 Hz) in the absence (left) and presence (right) of 100 µM carbamazepine (CBZ). (B) Time course of the peak amplitude of TTX-R Na+ currents during a train of 20 pulses (5 Hz) in the absence (open circles) and presence (closed circles) of 100 µM carbamazepine. Each point represents the mean and SEM from 11 experiments. (C) Carbamazepine-induced changes in the P20/P1 ratio of TTX-R Na+ currents (a, 1 Hz; b, 5 Hz). Each column represents the mean and SEM from 11 experiments. **p<0.01.
![]()
Effects of carbamazepine on inactivation kinetics of TTX-R Na+ channels
The inactivation kinetics of Na+ channels plays a crucial role in the generation of repetitive action potentials during a sustained depolarization. Therefore, we studied the effects of carbamazepine on the inactivation kinetics of TTX-R Na+ channels in medium-sized DiI-positive neurons with a two-pulse protocol. Conditioning pulses (P1) varying between 2 and 8000 ms in duration were followed after 20 ms by test pulses (P2), which elicited TTX-R Na+ currents (Figs. 6A and B). The P2/P1 ratios of TTX-R Na+ currents were used as parameters for channel inactivation. The P2/P1 ratios, when plotted against the durations of the conditioning pulses, were well fitted to a double exponential function (Fig. 6C). This yielded two time constants, a fast (τfast) and a slow (τslow) constant, suggesting that τfast and τslow represent time constants for the fast and slow component of channel inactivation. Carbamazepine increased changes in P2/P1 ratios with increased lengths of prepulse time (Fig. 6D). The application of 100 µM carbamazepine significantly decreased both τfast (control: 399.9±65.3 ms, carbamazepine: 68.7±10.9 ms, decrease by 76.0±7.4%, n=8, p<0.01) and τslow (control: 2055.1±415.9 ms, carbamazepine: 1223.7±205.0 ms, decrease by 37.5±5.8%, n=8, p<0.05) (Figs. 6Ea and Eb). In addition, τweighted was decreased from 1547.4±344.6 ms for the control to 844.3±131.4 ms for the carbamazepine condition (decrease by 42.1±4.2%, n=8, p<0.05, Fig. 6Ec), suggesting that carbamazepine accelerates inactivation during sustained membrane depolarization.
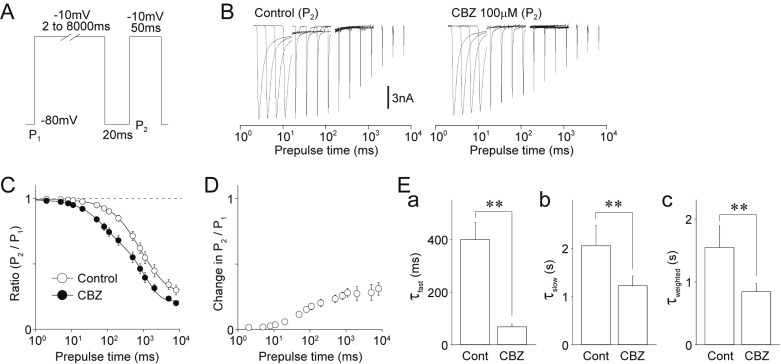
Fig. 6
Effects of carbamazepine on the onset of inactivation of TTX-R Na+ channels.
(A) A schematic illustration of voltage step pulses. The TTX-R Na+ currents were induced by a two-pulse protocol. Conditioning pulses (P1; −10 mV depolarization, 2–8000 ms duration) were followed by the second test pulses (P2; −10 mV depolarization, 50 ms duration). The second TTX-R Na+ currents were recovered with an interpulse interval of 20 ms at a potential of −80 mV. (B) Typical traces of TTX-R INa elicited by the second test pulses of the two-pulse protocol in the absence (left) and presence (right) of 100 µM carbamazepine (CBZ). (C) Kinetics for the recovery from inactivation of TTX-R Na+ channels in the absence (open circles) and presence (closed circles) of 100 µM carbamazepine. Each point represents the mean and SEM from 8 experiments. (D) Carbamazepine-induced changes in the P2/P1 ratio of TTX-R Na+ currents. Each point represents the mean and SEM from 8 experiments. (E) Carbamazepine-induced changes in the kinetic parameters [a, fast time constants (τfast); b, slow time constants (τslow); c, weighted time constants (τweighted)] for the recovery from inactivation of the TTX-R Na+ currents. Each column represents the mean and SEM from 8 experiments. **p<0.01.
![]()
Next, TTX-R Na+ currents were elicited by a conditioning pulse (P1, duration 500 ms), which was followed by a test pulse (P2) with varying interpulse intervals (1–5000 ms) at −80 mV (Fig. 7A and B). The P2/P1 ratios of TTX-R Na+ currents were used as kinetics parameters for the recovery from inactivation. The mean P2/P1 ratios of TTX-R Na+ currents were plotted against the interpulse intervals and fitted to a double exponential function (Fig. 7C). This yielded two time constants τfast and τslow, suggesting again that τfast and τslow represent time constants for the fast and slow recovery from inactivation, respectively. Carbamazepine-induced changes in the P2/P1 ratios were decreased with the length of recovery time from inactivation (Fig. 7D). Application of 100 µM carbamazepine significantly increased both τfast (control: 2.0±1.4 ms, carbamazepine: 6.9±1.5 ms, increase by 234.1±66.0%, n=10, p<0.05) and τweighted (control: 38.6±5.2 ms, carbamazepine: 52.5±4.9 ms, increase by 54.0±17.9%, n=10, p<0.01) (Figs. 7Ea and Ec). However, τslow was not affected by carbamazepine (control: 221.4±27.0 ms, carbamazepine: 233.1±25.2 ms, increase by 24.1±21.1%, n=10, p=0.40, Fig. 7Eb).
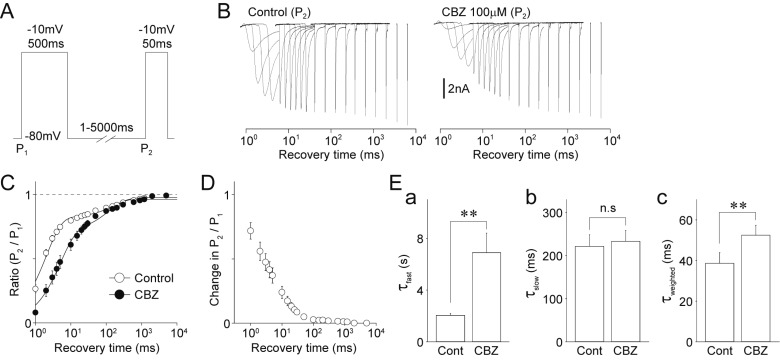
Fig. 7
Effects of carbamazepine on the recovery from inactivation of TTX-R Na+ channels.
(A) A schematic illustration of voltage step pulses. The TTX-R Na+ currents were induced by a two-pulse protocol. Conditioning pulses (P1; −10 mV depolarization, 500 ms duration) were followed by the second test pulses (P2; −10 mV depolarization, 50 ms duration). The second TTX-R Na+ currents were recovered with various interpulse intervals of 1 to 5000 ms at a potential of −80 mV. (B) Typical traces of TTX-R INa elicited by the second test pulses of the two-pulse protocol in the absence (left) and presence (right) of 100 µM carbamazepine (CBZ). (C) Kinetics for the recovery from inactivation of TTX-R Na+ channels in the absence (open circles) and presence (closed circles) of 100 µM carbamazepine. Each point represents the mean and SEM from 10 experiments. (D) Carbamazepine-induced changes in the P2/P1 ratio of TTX-R INa. Each point represents the mean and SEM from 10 experiments. (E) Carbamazepine-induced changes in the kinetic parameters [a, fast time constants (τfast); b, slow time constants (τslow); c, weighted time constants (τweighted)] for the recovery from inactivation of the TTX-R Na+ currents. Each column represents the mean and SEM from 10 experiments. **p<0.01; n.s, not significant.
![]()
Effect of carbamazepine on the excitability of dural afferent neurons
Finally, the effects of carbamazepine on the excitability of medium-sized DiI-positive neurons were examined in current-clamp experiments. In the presence of 300 nM TTX, the resting membrane potentials of medium-sized DiI-positive neurons ranged from −45.2 mV to −63.3 mV with a mean value of −54.2±5.9 mV (n=11, standard deviation). The rheobase currents, which represent the threshold currents (T) to generate action potentials, were typically between 100 pA and 300 pA (181.8±18.2 pA, n=11). In these current-clamp experiments, integers of rheobase current (1T to 4T) were injected to medium-sized DiI-positive neurons (Figs. 8A and B). Such depolarizing step current injections for a duration of 1000 ms generated multiple action potentials in most medium-sized DiI-positive neurons (8 of 11 neurons, 72.7%). In these conditions, 100 µM carbamazepine had no effect on the rheobase currents (122.7±14.1% of the control, n=8, p=0.19, Figs. 8B and C). However, carbamazepine significantly decreased the number of action potentials elicited by the depolarizing current injections, reducing the number of action potentials elicited at 4T to 49.3±17.9% of the control (n=8, p<0.05, Fig. 8C). In addition, carbamazepine (100 µM) increased the rheobase currents to 168.2±22.6% of the control (n=8, p<0.01, Fig. 8D). In addition, 100 µM carbamazepine had no effect on the voltage changes in response to hyperpolarizing current injections (Fig. 8E).
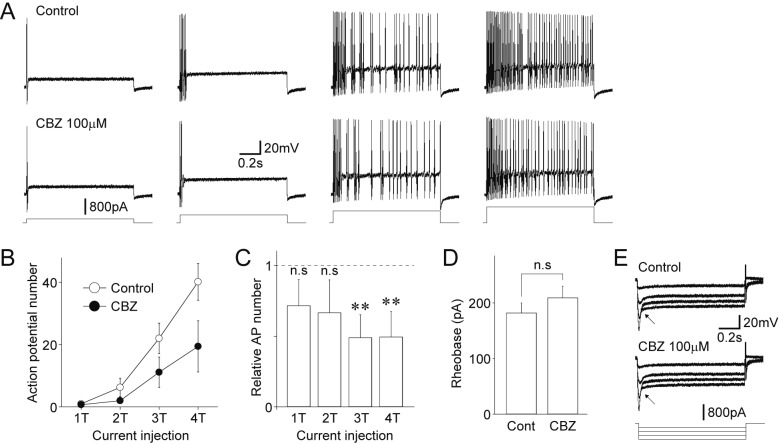
Fig. 8
Effects of carbamazepine on the excitability of dural afferent neurons.
(A) Effect of carbamazepine on voltage responses to depolarizing current injection in the absence (upper) and presence (lower) of 100 µM carbamazepine (CBZ). Four representative raw voltage traces were elicited by 1-fold threshold (1T; 200 pA) to 4-fold threshold (4T; 800 pA) depolarizing current injection. Note that the number of action potentials increases with stronger depolarizing current injection. (B) Changes in the number of action potentials elicited by depolarizing current injection (1T to 4T) in the absence (open circles) and presence (closed circles) of 100 µM carbamazepine. Each point represents the mean and SEM from 8 experiments. (C) Carbamazepine-induced changes in the number of action potentials elicited by depolarizing current injection (1T to 4T). Each column represents the mean and SEM from 8 experiments. **p<0.01, n.s; not significant. (D) Carbamazepine-induced changes in the amplitude of rheobase currents. Each column represents the mean and SEM from 8 experiments. n.s; not significant. (E) Typical voltage responses to hyperpolarizing current injection in the absence (upper) and presence (lower) of 100 µM carbamazepine. DiI-positive neurons were current-clamped and hyperpolarizing step pulses (−200 pA increments, up to −800 pA, 1 s duration) were applied to patched neurons. Note that the sag potentials (arrows), which are typical properties mediated by HCN channels, were not affected by adding carbamazepine.
![]()
DISCUSSION
The goal of preventive medications is a decrease in frequency as well as in severity of migraine attacks and related symptoms. It is now well established that many different groups of drugs, e.g., anticonvulsants (topiramate, valproic acid, gabapentin), antidepressants (amitriptyline, duloxetine, fluoxetine), β-adrenoceptor antagonists (propranolol, nadolol, metoprolol), Ca2+ channel antagonists (flunarizine), and other miscellaneous drugs are effective for prevention of migraine attacks [89]. Anticonvulsants targeting on voltage-gated Na+ channels have been investigated and used for migraine prevention, as abnormalities of these ion channels are implicated in epilepsy as well as in CSD [11]. Based on placebo-controlled studies, valproic acid and topiramate are the most effective anticonvulsants drugs to decrease the frequency and severity of migraine attacks [12132627]. Lamotrigine decreases the frequency of headaches and the duration of migraine attacks in patients with aura [28], but a placebo-controlled study has shown a lack of efficacy for lamotrigine in migraine prevention [15]. Zonisamide decreases the severity and frequency of migraine attacks similar to topiramate and can be considered as an alternative preventive medication [2930]. Oxcarbazepine, a structural derivative of carbamazepine, has been found to be not effective in the migraine prevention [16]. Although an early clinical study has shown the significant benefit of carbamazepine in the migraine prevention [14], further evidence is needed to determine whether this drug is effective in the prevention of migraine attacks.
Anticonvulsants act through a wide range of differential mechanisms of action, which ultimately modulate neural systems involved in the pathophysiology of migraine. Therefore, it is not easy to distinguish which mechanisms are responsible for their therapeutic effects. As discussed above, a subset of anticonvulsants inhibiting voltage-gated Na+ channels are effective in migraine prevention. This suggests that the modulation of these channels is not the only mechanism underlying migraine prevention by Na+ channel blockers. For example, valproic acid decreases brain levels of aspartate and glutamate, and inhibits GABA transaminase and activates glutamic acid decarboxylase leading to increased GABAergic inhibitory transmission [31]. In addition, topiramate is known to enhance GABA-evoked currents, to inhibit kainate-evoked currents, and to inhibit carbonic anhydrases [32]. Both carbamazepine and oxcarbazepine exert additional actions on voltage-gated Ca2+ channels [33], and carbamazepine is also known to increase serotonin concentration [343536]. The inhibition of serotonin and norepinephrine transporters is a major action mechanism of actions for tricyclic antidepressants like amitriptyline [37], which are widely used for migraine prevention [938]. This supports the idea that additional effects of carbamazepine on the serotonin system may contribute to its preventive efficacy against migraine attacks.
On the other hand, neurogenic inflammation triggered by vasoactive neuropeptides, such as calcitonin gene-related peptide and substance P, results in the vasodilation and generation of inflammatory mediators including histamine, serotonin, and prostaglandin E2 in the dura mater [3940]. Inflammatory mediators directly stimulate dural afferent neurons to transmit nociceptive information on to the central nervous system, or sensitize peripheral terminals of dural afferent neurons [241]. Importantly, such inflammatory mediators are known to potentiate TTX-R Na+ currents in nociceptive neurons [4243], suggesting the possible roles of TTX-R Na+ channels in migraine pathology. In the present study, we examined the effects of carbamazepine on TTX-R Na+ channels in medium-sized dural afferent neurons identified with the fluorescent dye DiI. These neurons belong to polymodal C-type nociceptive neurons, based on their sensitivity to acidic pH, αβ-me-ATP, and capsaicin [44]. The present study is the first description of a carbamazepine effect on TTX-R Na+ channels in dural afferent neurons. We found that carbamazepine had little effect on the peak amplitude of transient TTX-R Na+ currents (IC50 value of >1 mM). In contrast, we found that carbamazepine, even at concentrations below 100 µM, significantly decreased the amplitudes of steady-state component of TTX-R Na+ currents and slow voltage ramp-induced persistent Na+ currents with IC50 values of 165 µM and 71 µM, respectively. Given that therapeutic levels of carbamazepine during long-term treatment are in the range of 4–12 µg/ml (17–51 µM) [4546], carbamazepine may decrease the excitability of dural afferent neurons by inhibition of persistent rather than transient TTX-R Na+ currents. In lines with the present results, several anticonvulsants proven to be effective in the migraine prevention, especially valproic acid and topiramate, more potently inhibit the persistent than the fast transient Na+ currents [4748]. Although further studies are needed to reveal the relationship between the preferential blockade of persistent Na+ currents and preventive efficacy against migraine attacks, persistent Na+ currents are regarded as one of the pharmacological targets for the development of new drugs against epilepsy and neuropathic pain [4950].
We also found that carbamazepine had little effect on the use-dependent inhibition and voltage-activation relationship of TTX-R Na+ channels. These results are supported by previous studies showing the limited effect of carbamazepine on these properties of TTX-R Na+ channels [222351]. We also found that carbamazepine slightly but significantly shifted the voltage dependence of fast as well as slow inactivation of TTX-R Na+ channels towards hyperpolarized potentials, which is consistent with the preferential binding of carbamazepine to Na+ channels at depolarized membrane potentials [515253]. Additionally, the present results show the effects of carbamazepine on the onset of inactivation of TTX-R Na+ channels. Carbamazepine decreased both the τfast and τslow, suggesting that it accelerated both components of the inactivation kinetics in TTX-R Na+ channels. However, carbamazepine increased the τfast but not τslow in the recovery kinetics, suggesting that carbamazepine slows down the fast component of recovery from inactivation. A delayed fast recovery from inactivation of TTX-R Na+ channels might be responsible for a reduced number of action potentials during a sustained depolarization.
In the present study, medium-sized dural afferent neurons showed repetitive action potentials during a sustained membrane depolarization. Such a firing pattern in response to depolarizing current stimuli may result from subthreshold membrane conductances, which are mediated by persistent Na+ currents [54555657]. We found that carbamazepine had no effect on rheobase currents, suggesting that it does not change the threshold for the generation of action potentials mediated by TTX-R Na+ channels. In contrast, carbamazepine significantly reduced the number of action potentials elicited by moderate depolarizing current injections. Considering that carbamazepine preferentially inhibited non-inactivating and/or persistent TTX-R Na+ currents and that it delayed the recovery from inactivation of these channels, these modulations could be responsible for the carbamazepine-induced decrease in the excitability of dural afferent neurons. On the other hand, hyperpolarization-activated cyclic nucleotide-gated (HCN) channels also contribute to the regular and repetitive generation of action potentials in various central neurons [58]. However, an involvement of HCN channels in the carbamazepine-induced decrease in the excitability of dural afferent neurons is probably negligible, because carbamazepine had no effect on the sag potentials, which are generated by HCN channels.
In summary, we found that carbamazepine at clinically relevant concentrations inhibited the non-inactivating and/or persistent Na+ currents mediated by TTX-R Na+ channels in C-type dural afferent neurons. We also found that carbamazepine modulated the inactivation kinetics of TTX-R Na+ channels, resulting in decreased channel availability during sustained depolarization, thereby influencing the repeated generation of action potentials. Although the present results has provided clear evidence for the effects of carbamazepine on TTX-R Na+ channels in dural afferent neurons, clinical evidence is required to determine whether carbamazepine is effective in the prevention of migraine attacks.
 XML Download
XML Download