

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Obstructive sleep apnea (OSA) is characterized by repeated episodes of complete or partial upper airway collapse during sleep, causing intermittent hypoxia (IH), arousal, and sleep fragmentation.1 OSA has become a public health concern as it leads to clinical complications,2 including cardiovascular disease (CVD) as the most severe.3 Chronic obstructive pulmonary disease (COPD) is a disorder characterized by poorly reversible airflow limitation that is usually progressive and associated with an abnormal inflammatory response of lungs to noxious particles, particularly cigarette smoke.4 COPD occurs in about 11.4% of people aged ≥30 years globally, according to a recent meta-analysis,5 and it is estimated that between 30% and 50% of COPD-related deaths are due to cardiovascular comorbidities such as coronary artery disease, hypertension, or diabetes.6 Chronic hypoxia plays a pivotal role in the systematic inflammation of COPD,78 and the concurrence of OSA and COPD is termed “overlap syndrome (OS)”, with an incidence of approximately 1% of adults in the general population.9 Although patients affected by OS have a higher risk of CVD and all-cause mortality compared to patients with COPD or OSA alone,10 underlying mechanisms linking OS and CVD are not well understood. We hypothesized that OS-related systemic inflammation and cell apoptosis may play important roles by promoting cardiovascular dysfunction leading to CVD.
Both OSA and COPD are recognized as independent risks factors for atherosclerotic CVD.1112 It is well established that OSA-associated IH and chronic continuous hypoxia associated with COPD promote systemic and vascular inflammation through increased activity of nuclear factor (NF)-κB, thereby resulting in endothelial dysfunction.71314 Endothelial dysfunction is the earliest stage of atherosclerotic process, and can trigger cardiovascular events.12 Intracellular adhesion molecule-1 (ICAM-1) and vascular cellular adhesion molecule-1 (VCAM-1) play vital roles in this process by inducing leukocyte adhesion and transmigration to the subendothelial space, and thus serve as useful indicators of endothelial dysfunction and vascular inflammation, respectively.1516 Increased expression of adhesion molecules will enable the recruitment and infiltration of immune cells that contribute to lesion development.17
Integrity and functional activity of the endothelial monolayer plays a critical role in atherogenesis.1819 Apoptotic endothelial cells are rarely found in vessels that are free of vascular pathology. However, when afflicted with diseases, apoptotic circulating endothelial cells (CECs) have been reported to be an ex vivo indicator of vascular injury.20 Accumulating evidence have shown that many of the risk factors linked to endothelial dysfunction can be caused by endothelial apoptosis.1820 Extensive endothelial cell apoptosis results in loss of integrity of the endothelium, which subsequently contributes to the progress of hypoxia-induced CVD. The level of apoptotic CECs has been reported to be increased in patients with OSA or COPD, while continuous positive airway pressure (CPAP) treatment improves hypoxia and decreases apoptosis of endothelial cells.21
Tempol (4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl) is an antioxidant that functions as a superoxide dismutase mimic, which can penetrate cell membranes and react with both intra- and extracellular oxygen free radicals, resulting in the protection of endothelial function.22 Previous studies23 showed that tempol can have a protective effect on the atherosclerosis associated with metabolic syndrome by decreasing the extent of vascular inflammation. Furthermore, treatment of IH-exposed rats with tempol was shown to restore vascular reactivity.24 Therefore, it is reasonable to hypothesize that tempol may attenuate the endothelial injury induced by OS.
To gain a better understanding of the mechanistic link between OS and CVD in addition to determining the potential protective effects of antioxidant tempol as a therapeutic option, the present study was designed to explore whether the severity of vascular endothelial injury is increased by elevated apoptosis of CECs and inflammatory response in OS-induced rats, and to assess the intervention effect of antioxidant tempol on these effects.
MATERIALS AND METHODS
Animals
Sixty-six male Wistar rats weighing 160–175 g at 6 weeks of age (obtained from the Model Animal Center, Radiological Medicine Research Institute, Chinese Academy of Medical Science, Tianjin, China) were used in this study. After 1 week of environmental adaptation, rats were randomly allocated to six groups (n=11 each) according to exposure conditions as follows: normal control (NC) group (normal oxygen control), IH group (IH exposure), CH group (cigarette smoke exposure), OS group (cigarette smoke and IH exposure), OST group (OS group treated with tempol), and OSN group (OS group treated with NaCl). Rats had free access to food and water. Room was maintained at a temperature of 22℃ with 45% humidity to ensure the most suitable environment for rats' survival. This study was approved by the Tianjin Medical University Animal Care and Use Committee (Ethical No. IRB 2018-YX-043).
Cigarette smoke exposure
Cigarette smoke exposure was established according to a previously published protocol.25 In brief, rats were placed in a home-made smoking device (0.6 m3 with five poles on a side wall for ventilation, six rats per batch) and exposed to the smoke of 15 commercial unfiltered cigarettes (Daqianmen™, Shanghai, China) for 30 min twice daily, first in the morning (before 9 am) and then in the evening (after 5 pm), for 8 weeks continuously. The contents of tar and nicotine were 18 mg and 1 mg per cigarette, respectively, and the burning of five cigarettes simultaneously resulted in a 15% smog content (v/v) within housing chamber. Rats were confirmed to be not experiencing discomfort during cigarette smoke exposure.
IH exposure
As previously described,26 we used a gas control delivery system for IH exposure, which regulated flow (5 L/min) of pure nitrogen or clean air continuously into a customized IH housing chamber (23×20×12 cm=5520 cm3≈5.5 L) to maintain designated continuous hypoxia or normoxia environment. Software edited with Visual C++ computer language (Jing Feng, Tianjin Medical University General Hospital Respiratory Simulation System 1.0, Tianjin, China, 2005) was applied to control the duration of hypoxia (first 30 s) or reoxygenation (latter 90 s) phases, producing 30 cycles of alternations per hour. Subsequently, a series of programmable solenoids and flow regulators altered the fractional concentration of inspired oxygen. As a result, each chamber was filled with nitrogen for the first 30 s, so that the minimum oxygen concentration reached 5%, and then each chamber was filled with compressed air for the latter 90 s, so that oxygen concentration gradually returned to 20.9%. For OST and OSN groups, animals were treated with 100 mg of 10% (w/v) tempol or 0.9% NaCl per kilogram body weight by intraperitoneal injection under OS exposure conditions, respectively. Exposure experiments were performed from 9 am to 5 pm each day continuously for 8 weeks, corresponding to sleeping time for Wistar rats. Oxygen concentration monitors (Hamilton, Switzerland) were used to monitor changes in oxygen concentration and environment in the exposed container.
Control experiments were performed on animals exposed to alternating cycles of compressed room air in chamber.
Preliminary experiment to obtain arterial blood gas
Two days before the end of exposure period, three rats from each group were randomly selected for detection of arterial blood gas (ABG). After the rats were mildly anesthetized with 10% chloral hydrate (0.1 mL/100 g) intraperitoneally, local disinfectant was applied, and a microcatheter filled with heparin saline was inserted into right common carotid artery to achieve systemic heparinization, so as to prevent blood clotting during measurement of ABG. Rats were placed in oxygen chamber for 10 min, and arterial blood samples (0.7 mL each) were collected through capillary blood collection at different time points of hypoxia cycle. ABG analysis included measurements of partial oxygen pressure (PaO2), partial carbon dioxide pressure (PaCO2), arterial oxygen saturation (SaO2), and pH.
Blood collection
At the end of exposure period, the remaining eight rats of each group were anesthetized with 10% chloral hydrate (0.3 mL/100 g) intraperitoneally, and arterial blood sample was obtained from right femoral artery. Rats were killed by exsanguination. Serum was separated by centrifugation (Eppendorf, Germany) at 3000 rpm for 15 min and stored at −80℃ until analyzed. The other fresh blood sample was used for endothelial apoptosis detection by flow cytometry.
Histology of lung tissue
Lungs were removed, placed in 10% buffered formalin for at least 18 h, and then tissues were dried, immobilized, and embedded in paraffin. Tissue slices (5-µm diameter) were stained with hematoxylin and eosin. Iimages were captured at 40× magnification using an Olympus BX40 microscope (OLYMPUS, Tokyo, Japan). We made the following measurements to assess lung tissue according to a previous study.25 The objective measurements included mean linear intercept (MLI), mean alveolar number (MAN), and pathological score of lung inflammation.
Measurement of ICAM-1 and VCAM-1 levels
Serum levels of ICAM-1 and VCAM-1 were determined using enzyme-linked immunosorbent assay kits (R&D Systems, Inc., Minneapolis, MN, USA), according to the manufacturer's instructions. The optical density of each well was determined using a microplate reader (Thermo Science, Waltham, MA, USA) at 450 nm within 30 min.
Detection of apoptotic CECs
Peripheral blood mononuclear cells were isolated from rat peripheral blood by light-density separation using Histopaque 1083 (1.083 g/mL at 25℃) and Histopaque 1119 (1.119 g/mL at 25℃), according to the manufacturer's instructions. Cells were resuspended in 100 µL binding buffer with 5-µL Purified NA/LE Mouse Anti-Rat CD31 (BD Biosciences, San Diego, CA, USA) as the primary antibody, and incubated for 15 min in the dark at room temperature. Cells were then washed once with 1 mL of binding buffer, and pelleted by centrifugation at 1500 rpm for 5 min. Apoptosis of CECs was evaluated by resuspending cells in 150 µL Annexin V Binding Buffer and incubating them with 5-µL FITC Goat Anti-Mouse IgG/IgM, 5-µL 7-Amino Actinomycin D (7-AAD), and 5-µL PE-Annexin V (all from BD Biosciences) for 15 min. PE-Annexin V has high affinity for membrane phospholipid phosphatidylserine, and thus serves as a sensitive probe for flow cytometric analysis of cells that are undergoing apoptosis, whereas 7-AAD differentiates dead (7-AAD positive) from apoptotic (7-AAD negative) cells. After incubation, samples were analyzed by a FACS Calibur flow cytometer (BD Biosciences) to assess cells that were positive for CD31 and Annexin V (CD31+Annexin+) and negative for 7-AAD (7-AAD−), and rate of endothelial apoptosis was measured according to the manufacturer's instructions. A total of 50000 events were measured per sample. On the basis of peripheral blood mononuclear cell counts, we calculated the absolute number of circulating CD31+AnnV+7-AAD− cells, expressed as a percentage of total cell count.
Statistical analysis
All data analyses were performed using SPSS version 16.0 software (SPSS Inc., Chicago, IL, USA). All data are expressed as means±SD. Preliminary ABG data were obtained and displayed as descriptive statistics only, but were not further analyzed for significant differences. For other variables, group differences were analyzed by one-way ANOVA, followed by Bonferroni post-hoc test for multiple comparisons. p values <0.05 were considered statistically significant.
RESULTS
Oxygen concentration in exposure container
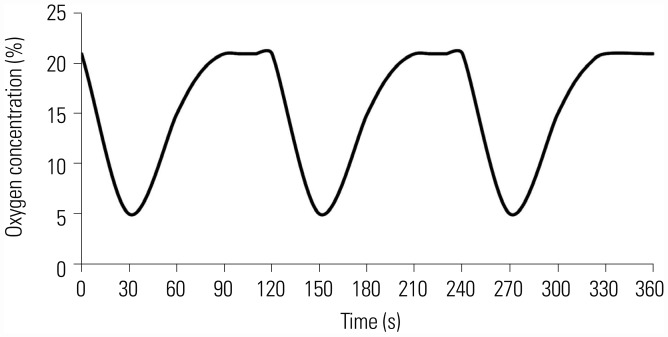
Fig. 1 shows oxygen concentration of chamber monitored in real time by an oxygen concentration monitor. In hypoxic stage (the first 30 s), oxygen concentration gradually decreased to the minimum of 5%, and then gradually increased to 21% in reoxygenation stage (latter 90 s).
Arterial blood gas
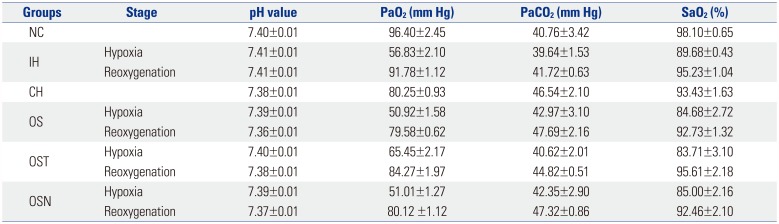
As shown in Table 1, PaO2 value of NC group, which was maintained in the normal physiological state, was greater than that of IH, OS, OSN, and OST groups in both hypoxia and reoxygenation stages. Although PaO2 values increased in reoxygenation stage in exposure groups, they fluctuated throughout the experimental period.
Pathological changes in lungs Histological images of lung sections
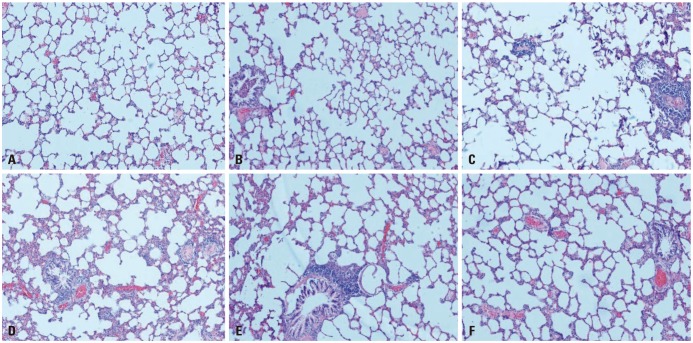
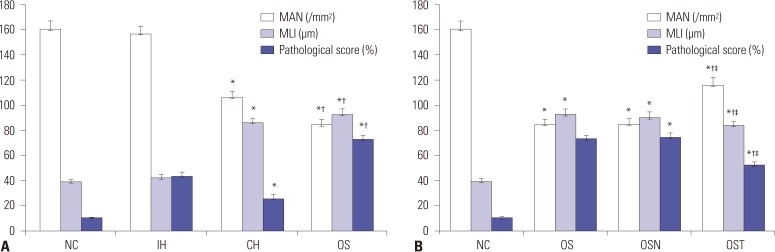
Histological images of lung sections randomly selected from each group are presented in Fig. 2. Lung tissues in NC group appeared normal under the light microscope, and alveolar size and distribution were relatively average (Fig. 2A). In IH group, inflammatory cells infiltration of some vascular walls, and lymphocytic infiltration of tracheal wall and interstitial lung were observed (Fig. 2B). Pathological changes in lungs of CH group (Fig. 2C) included the development of emphysema accompanied by infiltration of inflammatory cells, partial fracture of bronchial wall smooth muscle, and goblet cell hyperplasia, which were more severe in OS and OSN groups (Fig. 2D and E). Therefore, smoke exposure exacerbated the inflammation caused by IH and emphysema, which was notably reduced in lung tissues from OST group (Fig. 2F). Compared to NC or IH group, MAN was significantly decreased and MLI was increased in CH or OS group (all p<0.001). Moreover, pathological scores were higher in IH or OS group than in CH or NC group. In addition, the intervention of tempol reversed these effects by increasing MAN and decreasing MLI or pathological scores (all p<0.001) (Fig. 3).
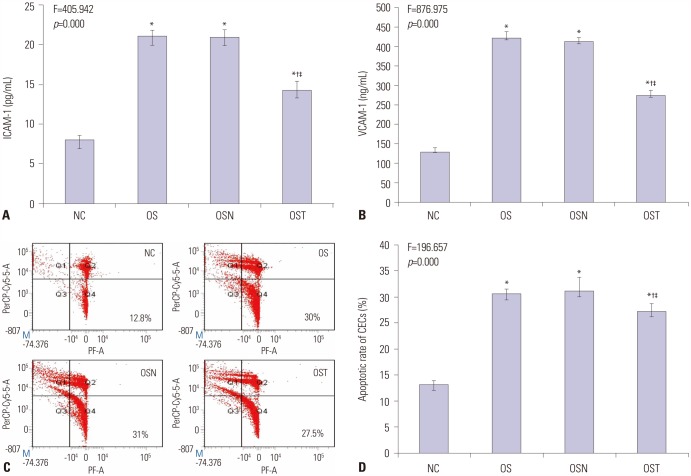
Serum concentrations of ICAM-1 and VCAM-1
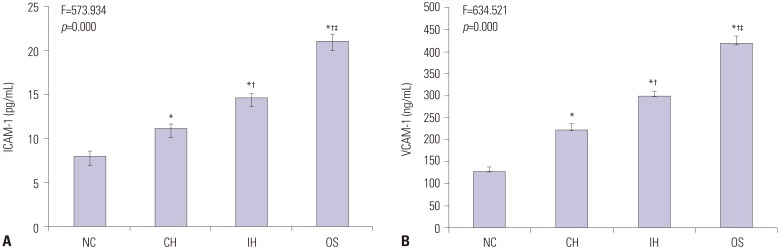
Quantification of serum levels of cell adhesion molecules are summarized in Supplementary Table 1 (only online). As presented in Fig. 4, serum concentrations of ICAM-1 and VCAM-1 were significantly increased in OS group compared to those in NC, CH, and IH groups (all p<0.001). Serum concentrations of both factors were also increased in CH and IH groups compared to those of NC group (all p<0.001). Moreover, both ICAM-1 and VCAM-1 serum concentrations were higher in IH group than in CH group (p<0.001).
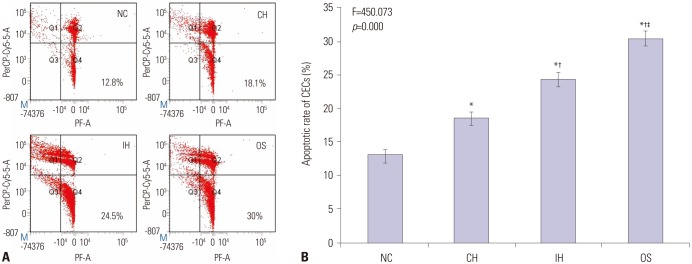
Apoptosis of CECs
The distribution of CECs in NC, IH, CH, and OS groups were observed by flow cytometric assays, from which apoptotic cells can be distinguished from healthy and dead cells (Fig. 5A). As shown in Fig. 5B, highest apoptotic rate was observed in OS group, and lowest was observed in NC group (all p<0.001). In addition, apoptotic rate of CECs in IH group was higher than that in CH group (p<0.001). Quantification of CECs apoptosis rates is summarized in Supplementary Table 2 (only online).
Effect of antioxidant tempol on serum ICAM-1, VCAM-1, and CECs apoptosis
As shown in Fig. 6A and B, and Supplementary Table 3 (only online), serum ICAM-1 and VCAM-1 levels in OST group were higher than those in NC group, and were lower than those in OS and OSN groups (p<0.001), with no significant differences between OS and OSN groups (p>0.05). As shown in Fig. 6C and D, tempol also reversed these effects, in which CECs apoptosis rate in OST group was higher than that in NC group, and was lower than those in OS and OSN groups (p<0.001) (Supplementary Table 4, only online).
DISCUSSION
We demonstrated that the inflammatory response and endothelial apoptosis involved in pathophysiological endothelial dysfunction induced by IH and smoke exposure were significantly elevated in a rat model of OS produced through IH exposure during sleep, together with emphysema, during 8 weeks of smoke exposure. More importantly, we found that treatment with antioxidant tempol provided a partial protective effect against induced endothelial injury.
ABG results obtained from preliminary experiments and pathological characteristics of rats exposed to smoke confirmed successful establishment of the rat model of OS by inducing IH together with emphysema due to cigarette smoke exposure, which met the pathological criteria.27 Emphysema is a main feature of COPD and plays a critical role in its pathophysiology. Emphysematous models induced by smoke have become a focal point in COPD research. Such models do not fully simulate pathological features of COPD; however, they remain ideal at present. The longer rats are exposed, the more easily the model is established. A previous study in China confirmed that rats showed emphysematous changes after 7 weeks of smoke exposure.28 We observed early emphysema (including alveolar wall rupture, alveolar enlargement, infiltration of inflammatory cells, MLI increases, and MAN decreases) after 8 weeks. Therefore, the emphysematous model was established successfully according to pathological results. Many OSA studies use IH as the only exposure factor. Our acute model further allowed us to evaluate the early effects of these injurious stimuli while avoiding other confounding factors found in OSA patients which also cause inflammation and endothelial dysfunction, such as metabolic syndrome, obesity, and hypertension.29 However, this model ignores anatomical site of upper airway obstruction, and does not take into account the effects of animal sleep and arousal on blood oxygen levels. Nevertheless, these factors would not influence the primary focus of our study. Moreover, ABG analysis is an important basis for assessing hypoxia of IH animals and OSA patients. Therefore, ABG was used to evaluate hypoxic changes of IH chamber, as well as to confirm the validity and rationality of the model.
Several studies have suggested that the inflammatory processes associated with endothelial dysfunction plays key roles in the development of cardiovascular complications in patients with COPD and OSA.1430 ICAM-1 and VCAM-1 are two important cell adhesion molecules that are ubiquitous in low concentrations in membranes of leukocytes and endothelial cells. Therefore, CAMs are good indicators of endothelial dysfunction and vascular inflammation.31 The adherence of monocytes to vascular endothelial cells and their transformation into macrophages is a critical point in atherosclerosis prophase,1 and ICAM-1 and VCAM-1 play vital roles in this process. Previous studies323334 have shown that these molecules are elevated in patients with OSA or COPD. We also revealed enhanced levels of ICAM-1 and VCAM-1 in CH and IH groups compared to those of NC group, supporting an increase in the systemic inflammation of rats after exposure to cigarette smoke or IH. We also found that the levels of ICAM-1 and VCAM-1 in rats exposed to IH were significantly higher than those of rats exposed to cigarette smoke alone, suggesting that IH may lead to a more severe inflammatory response compared to emphysema. An adaptive response in IH may not work well, due to the initiation of oxidative stress; as a result, with prolonged exposure, there will be an increase in anti-inflammatory mechanisms and a decrease in the impaired mechanism, resulting in a counterbalance between protection and injury. Therefore, differences in the inflammatory response of rats exposed to IH for different periods of time should be further investigated. Despite these nuances, we clearly demonstrated that serum ICAM-1 and VCAM-1 concentrations were the highest in OS group. This indicates that both IH and cigarette smoke exposure, overlapped either directly or synergistically, may elicit a more severe systematic inflammatory response than either stimulus alone. Our result is consistent with a previous study25 that assessed systematic inflammation, oxidative stress, and hypercoagulability in emphysematous rats exposed to IH. Therefore, increased CAM levels can increase the risk for future cardiovascular events.
Endothelial cells are the innermost layer of blood vessels and play an important role in blood vessel integrity and function, in which injury and apoptosis are the most common pathogenic mechanisms of multiple cardiovascular system diseases, such as arteriosclerosis and thrombus.35 Detection of endothelial injury is a relatively new method to directly determine the number of CECs. Previous studies720 have implied that apoptosis of endothelial cells is also a main influencing factor underlying endothelial dysfunction in OSA and COPD. In addition, patients with OSA have increased density of apoptotic CECs compared to non-OSA subjects, and the levels of apoptotic endothelial cells are correlated with abnormal endothelial vasorelaxation.36 CPAP therapy reduces the level of apoptotic endothelial cells. Cigarette smoke can also induce excessive apoptosis in endothelial cells,37 which was confirmed by the present study in which apoptosis rate was significantly increased by IH or smoking exposure. Similar findings were observed in patients diagnosed with acute coronary syndrome.38 Moreover, apoptosis rate of CECs in IH group was significantly higher that than of CH group, indicating aggravated endothelial injury. Importantly, OS group showed the highest level of CEC apoptosis, which implies the same state in patients with OS, where the combination of COPD and OSA may induce worse vascular injury than in patients who only have only one of the diseases.
Tempol is a redox-cycling nitroxyl radical that promotes the scavenging of many reactive oxygen species, and acts as an antioxidant through increasing superoxide dismutase activity.39 Tempol treatment has been reported to attenuate the IH-mediated increases of inflammatory factors such as tumor necrosis factor-alpha, ICAM-1, and NF-kβ. Early tempol intervention was also found to be more beneficial than tempol treatment initiated at a later time point.40 Tempol was also found to attenuate the pro-apoptotic effect of IH-induced lymphocytes on endothelial cells.26 We also demonstrated a significant decrease in the apoptosis level of endothelial cells and adhesion molecule levels in OS rats treated with tempol. These results may support the potential of tempol as a treatment for OS-induced cardiovascular complications as an alternative to CPAP. However, the above indicators in rats treated with tempol were still higher than those in NC group, indicating that tempol could only partly improve endothelial injury when IH is combined with emphysema.
This study has some limitations. First, we did not detect apoptosis of polymorphonuclear neutrophils and pathological changes of cardiovascular system. The crosstalk between endothelial cells and circulating inflammatory cells is an important aspect contributing to vascular injury, which will provide stronger evidence of endothelial dysfunction. Second, mechanisms of endothelial apoptosis were not evaluated. Although previous studies have reported the role of cell death receptors and mitochondria-dependent apoptotic pathways in this process, it is important to clarify the pathway involved in apoptosis of endothelial cells for establishing future intervention targets. Third, owing to a lack of equipment for measuring lung function in rats, we could not directly prove that airflow was limited after 8 weeks of smoke exposure. Finally, we did not evaluate the changes in blood pressure and cardiac factors (e.g., left ventricular hypertrophy) in rats, which are also related to endothelial cell dysfunction.
In summary, using an OS rat model, we detected synergistic effects when IH is combined with emphysema induced by smoke, resulting in more severe inflammation and endothelial apoptosis. This implies that patients with OS may be at an elevated risk of cardiovascular health problems. Moreover, treatment with antioxidants, such as tempol, could provide at least a partially protective effect against endothelial injury by suppressing the inflammatory response and endothelial apoptosis. Given the significant clinical implications of these results, these findings warrant further investigation in humans.
 XML Download
XML Download