

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Long-term intense exercise (IE) induced myocardial fibrosis (MF) has been characterized in both animal1)2) and human studies.3)4) However, not all human studies demonstrated this phenomenon,5)6) making the relationship between long-term intense endurance exercise and the development of MF unclear. Furthermore, studies observed MF with different patterns and location in lifelong endurance athletes.7) MF is generally associated with cardiac damage and repair, often characterized by inflammation.8) Although myocardial damage biomarkers were detected following IE,9) this process was suggested as a physical adaptation rather than pathological.10) Heidbuchel et al.11) suggested that long-term IE induced MF was associated with cardiac damage, but without supporting evidence. Thus, the basis for the association between IE induced cardiac damage and MF is poorly understood. Importantly, cardiac damage biomarkers including cardiac troponin and B-type natriuretic peptide (BNP) were detected after IE, but the exact origin is still unknown. Growing evidence has also demonstrated the right ventricle (RV) is vulnerable to IE.12) The aim of this study is to investigate whether serum troponin released by the RV results from IE induced myocardial damage.
Extreme exercise, both in human13) and animal studies,14) was shown to induce cardiac damage associated with acute inflammatory myocardial infiltrates. However, few studies investigated myocardial damage and inflammation after long-term IE, so the contribution of inflammation to exercise induced right ventricular fibrosis is unknown. In this study, we demonstrated that IE induced myocardial damage and following inflammation and fibrosis occurred exclusively in the RV, which is essential to the development of exercise induced MF.
Go to : 
METHODS
Animals
Adult male Sprague-Dawley (SD) rats (aged 2 months with similar weight 220±8 g) were purchased from Vital River Laboratories (VRL). The animals were housed in the specific pathogen free animal facility at the China Institute of Sport Science with free access to water and chow under a temperature and humidity controlled environment (room temperature 26±2°C; light/dark cycle 12/12 hours; relative humidity 40–45%). All animal procedures were in accordance with protocols approved by the national rodent care standards.
Exercise protocol
Animals were randomly assigned to sedentary (Sed), moderate exercise (ME), and intense exercise (IE) groups with varying durations (8,12, and 16 weeks). Thus, there were 9 groups total (n=8). The exercise program was based on a previously validated protocol.15) Exercise rats underwent treadmill running sessions 5 days/week. The protocol included a one-week progressive training program, starting with a 15-minute running session at 15 m/min and increasing gradually to steady-state 60-minute sessions at 28 m/min with 10° slope (81.0±3.5% VO2 max) for IE rats, and 60-minute sessions at 15 m/min with 5° slope for ME rats. Thereafter, animals were trained at respective level 5 days a week for 8, 12, or 16 weeks. Investigators observed the treadmill sessions daily to ensure effective running. Sed rats were housed and fed in the same conditions. Animals were euthanized 24 hours after the end of the training program, hearts were quickly removed; weighed; dissected into left ventricle (LV), RV, left atrium (LA), and right atrium (RA); and frozen in liquid nitrogen at −80°C or fixed for histological studies.
Detection of MF
The LV and RV were prepared in paraffin and sliced 4 μm thick, then treated with picric acid and Sirius red for collagen staining. Photographs were captured via the microscope camera system and analyzed with Image Pro plus 6.0 (Media Cybernetics, Inc., Silver Spring, MD, USA). The severity of myocardial interstitial fibrosis was determined (area of myocardial interstitial collagen/area of total field and perivascular collagen volume area [PVCA]/vascular area [VA]).
Serum cardiac troponin I (cTnI)
Rat cTnI (serum) enzyme-linked immunosorbent assay (ELISA) Kit (Life Diagnostics, West Chester, PA, USA) was used for the measurement of Serum cTnI concentration.
Observation of myocardial damage
Hematoxylin and eosin (H&E) staining: Paraffin sections of LV and RV were prepared, sliced (4-μm thick) and processed for H&E staining.
Transmission electron microscopy: LV and RV were immediately fixed in 2.5% glutaraldehyde and post-fixed in 1% osmium tetroxide. After ethanol gradient dehydration, samples were embedded in Epon 618 (TAAB Laboratories Equipment, Berks, UK). Ultrathin sections (50–70 nm) were cut and stained with uranyl acetate and lead citrate. Sections were then observed under the transmission electron microscope (HITACHI H-7650; Hitachi Ltd., Tokyo, Japan).
Western blot analysis
Protein expression of pro-inflammation factor interleukin (IL)-1β and monocyte chemotactic protein (MCP)-1 were examined in both ventricles. LV and RV sections were isolated and frozen in liquid nitrogen. Protein was extracted in a cell lysis buffer consisting of 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM Na2 ethylenediaminetetraacetic acid, 1 mM ethylene glycol tetraacetic acid, 1% Triton, 50 mM NaF, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, 2 mM benzamidine, 1 mg/mL leupeptin. After centrifugation for 15 minutes. at 12,000 ×g, supernatants were extracted, and protein concentration was determined using BCA Protein Assay Kit (Beyotime Institute of Biotech, Jiangsu, China). Protein was boiled at 95°C for 5 minutes in 5-fold concentrated sodium dodecyl sulfate (SDS) sample buffer (Beyotime Institute of Biotech). Eighty micrograms of each sample were loaded onto 12% polyacrylamide gel. After electrophoretic separation by SDS-polyacrylamide gel electrophoresis, the proteins were transferred to PVDF membranes (Immobilon-P; Millipore, Bedford, MA, USA). After the transfer process, the membranes were blocked in 5% non-fat milk (for glyceraldehyde 3-phosphate dehydrogenase [GAPDH], IL-1β, and MCP-1) in Tris-buffered saline with 0.1% Tween 20 (TBST) for 1.5 hours at room temperature. After blocking, membranes were incubated with the corresponding primary antibody (GAPDH, IL-1β, and MCP-1) in TBST with 5% non-fat milk at 4°C overnight. After the incubation, TBST-treated membranes were washed in TBST 4 times, 5 minutes per wash. After washing, the membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies at room temperature for 1 hour (1:10,000 dilution, 7074 for anti-rabbit IgG antibody; Cell Signaling Technology, Inc., Danvers, MA, USA). Blots were visualized using Millipore ECL according to the manufacturer's instructions and the protein bands were quantitatively analyzed by using an image analysis system (Image J software; National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Data were expressed as mean±standard error of the mean. Statistical analysis was generally carried out with one-way analysis of variance for the same time point. SPSS version 20.0 was used for statistical analysis (SPSS Inc., Chicago, IL, USA). Two-tailed values of p<0.05 were considered significant.
Go to :
RESULTS
Exercise induced right ventricular fibrosis
Rats were trained for 8, 12, and 16 weeks under ME or IE conditions with time matched Sed controls. After the administration of training, right ventricular collagen volume fraction (CVF), measured by means of picrosirius red staining, was significantly higher in intensely exercised rats compared to Sed rats (Figure 1). Furthermore, IE induced right ventricular fibrosis is time-dependent, with CVF increasing as the training duration increased. However, no differences were detected in left ventricular CVF among rats.
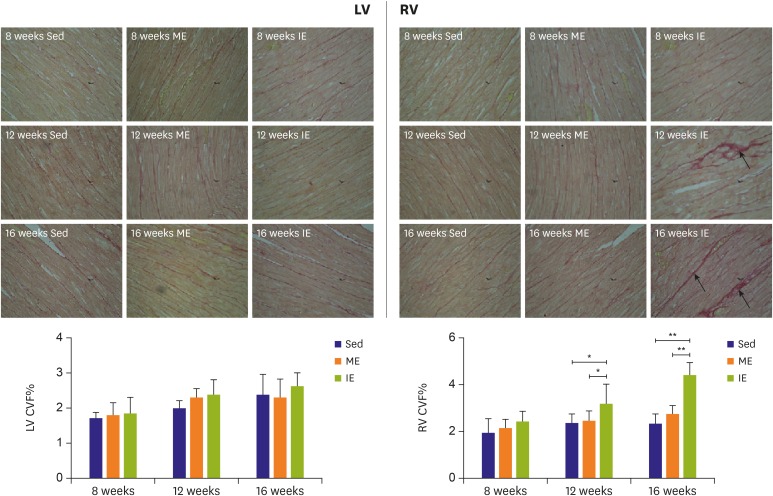 | Figure 1Picrosirius red staining (magnification ×400) and CVF of biventricular. MF occurred in RV rather than LV.CVF = collagen volume fraction; IE = intensive exercise; LV = left ventricle; ME = moderate exercise; MF = myocardial fibrosis; RV = right ventricle; Sed = sedentary.
*p<0.05; **p<0.01.
|
Right ventricular damage following IE
The primary purpose of our study was to determine the underlying mechanism of exercise induced MF, due to myocardial damage showing an association with this condition. To assess this, serum cTnI levels were measured. In line with previous studies, serum cTnI increased after IE compared to the Sed controls. Whereas, no significant difference was observed between the ME group and Sed group (Figure 2). However, the source of the increased serum cTnI following exercise was still unknown. To further determine the origin of the increased serum cTnI, the ultrastructure of the LV and RV were examined by transmission electron microscope. In intensely exercised rats, right ventricular fibers appeared to be abnormally distorted and the Z line blurred, however, left ventricular fibers remained neatly arranged (Figure 3). Thus, serum cTnI may come from damage to the RV. In contrast to Sed and moderately exercised rats, myofiber disarray and sporadic fragmentation of myocardial fibers were observed in the biventricular myocardium of IE rats. Interestingly, leukocyte infiltration was also exclusively observed in the RV (Figure 4).
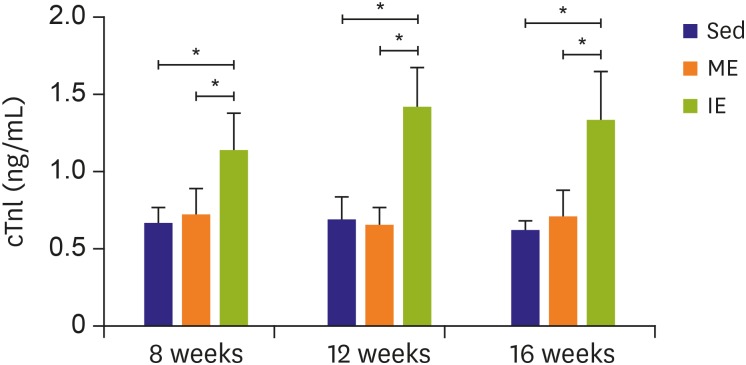 | Figure 2Serum cTnI. Serum cTnI level increased after IE training.cTnI = cardiac troponin I; IE = intensive exercise; ME = moderate exercise; Sed = sedentary.
**p<0.01.
|
 | Figure 3Biventricular myocardial cells ultra-structural changes (magnification ×2,000). In the RV of IE rats: the number of mitochondria increased and mitochondria size showed heterogeneity; fibers appeared to be abnormally distorted and Z line blurred.IE = intensive exercise; LV = left ventricle; ME = moderate exercise; RV = right ventricle; Sed = sedentary.
|
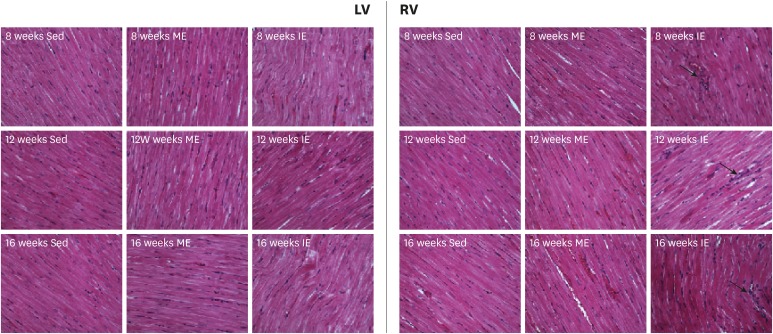 | Figure 4Histological alterations of biventricular (H&E, magnification ×400). Myocyte in exercise (both ME and IE) rats are thicker than Sed rats. Myofibre disarray and disrupt existed in RV of IE rats. Occasional mononuclear inflammatory cells infiltration (arrow) showed in RV of 12 weeks and 16 weeks IE rats.H&E = hematoxylin and eosin; IE = intensive exercise; LV = left ventricle; ME = moderate exercise; RV = right ventricle; Sed = sedentary.
|
Furthermore, a significant positive correlation was observed between right ventricular CVF and serum cTnI (r=0.597; p<0.001, Figure 5), but no correlation was found between left ventricular CVF and serum cTnI (p>0.05).

Inflammatory response in RV following IE
To more directly assess the involvement of inflammation in exercise induced MF, pro-inflammation factors IL-1β, and MCP-1 were analyzed (Figure 6). After IE, both IL-1β and MCP-1 protein expression level were increased in the RV when compared with the Sed group, while no difference was observed between the ME group and Sed group. However, the expression level of IL-1β and MCP-1 protein in LV was unchanged after long-term IE or ME.
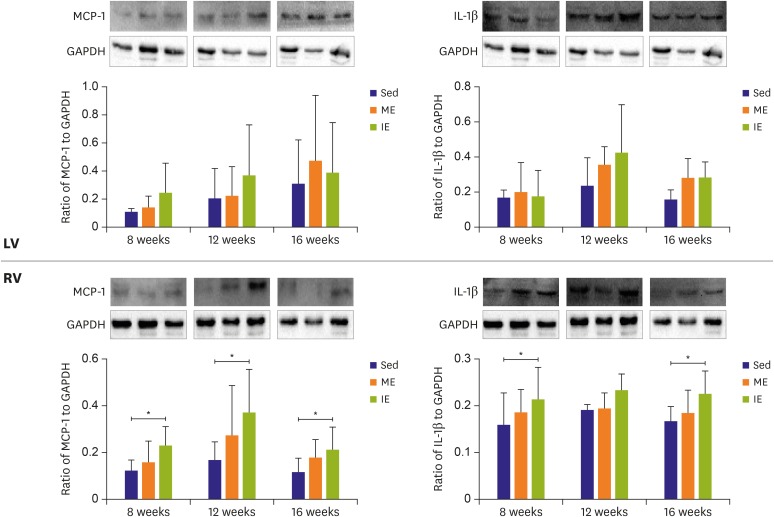 | Figure 6Proteins extracted from biventricular were assayed for levels of MCP-1 and IL-1β immunoblots, and immunoreactivating bands were quantified by densitometric analysis. Inflammation response was detected in RV rather than LV.GAPDH = glyceraldehyde 3-phosphate dehydrogenase; IE = intensive exercise; IL = interleukin; LV = left ventricle; MCP = monocyte chemotactic protein; ME = moderate exercise; RV = right ventricle; Sed = sedentary.
*p<0.05.
|
Go to :
DISCUSSION
Our results support the conclusion that long-term IE leads to right ventricular fibrosis. Despite numerous studies reporting that intensive endurance exercise would result in MF both in human3)16) and animal models,1)17) a bulk of the studies6)18) showed no MF in intensive endurance athletes. Thus, the relationship between long-term IE and the development of MF are still unknown. Van de Schoor et al.7) reviewed the potential mechanisms of MF in athletes, which include, genetic predisposition, silent myocarditis, pulmonary artery pressure overload, and prolonged exercise induced repetitive micro-injury were all noted as potential contributions. They also observed that the location of MF in studies varies substantially. While a previous animal study1) demonstrated that MF was observed in the RV, but remained absent in the LV. This suggests that there are many uncontrolled factors in human studies when investigating the relationship between IE and MF. Furthermore, most studies4) concluded exercise induced MF were based on cardiac magnetic resonance imaging (cMRI) data, which had been shown to have late gadolinium enhancement (LGE) in endurance athletes' heart. However, some investigators doubt the accuracy of LGE as a representation of MF, because LGE also could be found at the ventricular insertion point right after vigorous exercise, which may represent edema rather than fibrosis.19) Histology provides the only direct evidence of MF, but for human studies, samples are inaccessible and come with significant risks.
Therefore, in this animal study, picrosirius red staining was used to show the CVF in ventricles, which directly reflects tissue fibrosis. In line with previous research, our results demonstrated MF exclusively in the RV of IE rats. This suggests that RV is rendered more susceptive to form fibrosis after long-term IE. Tail shocks were involved in Benito's training protocol to stimulate continuous running,1) thus some researchers suggested that the stress response possibly is the cause of MF rather than exercise response. It should be noted that our exercise programs also were involuntary, but the electrical shock was not used to stimulate running, a quick hand touch was all that was needed to motivate our rats to continue running. RV fibrosis was still observed in our exercise protocol, thus we believe the stress response is not a major factor in the development of exercise-induced MF. Despite the similar RV fibrosis in our exercised rats, the relationship between fibrosis quantity and training time should be mentioned. In Benito and his colleagues' study,1) no progressive increases in fibrosis were found among 4, 8, and 16 weeks of training. Our results demonstrated that exercise-induced fibrosis is time-dependent, as the duration increases the fibrosis becomes more severe. This result further supports the idea that the exercise response is the main cause of RV fibrosis. Some researchers questioned why exercise induced fibrosis occurred only in the RV, but there was no hint of fibrosis in the LV.20) However, in another study, Aschar-Sobbi et al.17) demonstrated that MF occurred in the atria rather than LV following IE. The different alternations between RV and LV in long-term intense exercised animals motivated us to examine the underlying mechanism of the formation of exercise induced MF.
No MF was observed in the RV or LV following ME. This suggests that exercise intensity is central to the formation of MF. Exercise frequency is another essential factor to heart remodeling, it is reasonable to hypothesize exercise frequency is associated with MF. Unfortunately, we did not detect the role of exercise frequency on MF. Our study used the same frequency across all groups, thus we do not know what will happen if the rats had more recovery time between exercise bouts. Future studies investigating the effects of exercise frequency on MF are needed, which will make the relationship between IE and MF clearer.
There are still many unanswered significant questions in regard to the relationship between IE and MF. Among these questions we are most interested in why some athletes suffer MF whilst others not, which contrasts with animal studies were MF was detected in every IE group. For this question, we have our hypotheses as stated below. Firstly, it may be that genetic predisposition that influences MF formation. As reviewed by van de Schoor et al.7) and La Gerche,21) genetic predisposition is a potential factor for the development of MF. We believe that not only genetic mutations but also genetic diversity in individuals contribute to the variability of MF in athletes. However, in animal studies, all the animals nearly have the same genetic characteristics. Secondly, MF was detected in veteran endurance athletes rather than younger athletes, thus “age” may be a critical factor for the development of MF in athletes. Age here is not simply the age of the athlete, but also could also represent the “age” when the athlete begins training and the “age” of training history. Although the exact role of age on MF in athletes is unclear, age does distinctly play a role in human studies. While in animal studies each animal is normally age matched. Thirdly, training protocols including sport type, training frequency, and exercise intensity were entirely different in human studies, while animals were trained under the same conditions. Finally, different statistics and detection approaches are used on MF between human and animal studies. As previously discussed, human MF was detected by cMRI, while animal MF was detected by picrosirius red staining. All these hypotheses are plausible but mostly unsubstantiated, future studies are needed to confirm these hypotheses.
As seen in our intense exercised rats, myocardial damage occurred along with right ventricular fibrosis. In almost all studies on humans and animals following IE training, acute increases in cTnI and BNP have been detected.22) These biomarkers were used to serologically identify myocardial damage, which was well reviewed by Eijsvogels et al.23) In accordance with previous studies, our results also demonstrated that serum cTnI increased after IE. Importantly, these biomarkers represent myocardial damage, but to the best of our knowledge, where this damage occurs in the heart is still unknown. Therefore, we observed the morphology of the RV and LV under the ultrastructure level. Our results suggested that the source of the myocardial damage occurred in the RV rather than LV after long-term IE. Thus, increased serum cTnI is probably derived from right ventricular damage. In fact, most evidence so far suggests that RV function was reduced post-exercise.24)25) Also, studies reported moderately strong correlations between measures of RV dysfunction and levels of cardiac troponin and BNP.16) Thus, the RV is susceptive to IE induced impair. Although mechanisms are unclear, La Gerche et al.25) documented several potential reasons why the RV is more vulnerable. To summarize, IE resulted in an increase in wall stress of 125% vs. 4% for the RV and LV, the thin-walled RV possibly facilitates the progression due to increased wall stress causing more myocardial damage compared to the LV. Therefore, if the heart works under a profound and frequent exercise stimulus, without sufficient recovery, RV damage is possible.
In addition, based on different remodeling processes of the RV and LV following IE training, we hypothesis there is another potential mechanism of exercise induced RV damage. Arbab-Zadeh and his colleagues26) demonstrated that Sed individuals who intensely exercised for 12 months such as to finish a marathon; during the first 6–9 months LV showed concentric remodeling then returned to a normal mass to volume ratio thereafter, whereas the RV showed an eccentric remodeling throughout the whole program. This means that there are 2 stages of left ventricular remodeling. The first stage the LV becomes thicker, and then the LV becomes larger during the second stage. Therefore, the first stage increases ventricular myocardial fibers strength to meet the challenge of strenuous stretch during IE. While there is only one stage for right ventricular remodeling, which is becoming larger with no increase in thickness. It means that right ventricular myocardial fibers are not structurally ready to strenuously stretch, which results in right ventricular damage.
Furthermore, our study also uncovered moderately strong positive correlations between cTnI and right ventricular fibrosis. To our knowledge, our study is the first to successfully correlate serum cTnI and myocardial CVF, demonstrating a relationship between myocardial damage and fibrosis following long-term IE. However, there is little data to suggest that there is a pathological-biological pathway to link exercise induced myocardial damage and fibrosis. In contrast, many studies suggest that IE induced myocardial damage typically reverses within 24–48 hours of recovery and will not result in right ventricular dysfunction,27) thus exercise induced myocardial remodeling is physiological rather than pathological.10) Our study does suggest that exercise induced right ventricular damage is associated with right ventricular fibrosis, and there is a pathological pathway to link those 2 processes.
The relationship between exercise and inflammatory cytokines is complex. Low to moderate exercise has several established health benefits, and it has been suggested that anti-inflammation cytokines may potentiate some of these benefits.28) In animal studies, exhaustive exercise was shown to increase myocardial damage that was associated with inflammatory myocardial infiltrates.29) Human studies have displayed that pro-inflammatory cytokines increased after intense endurance exercise, and researchers suggested that this was possibly associated with cardiac dysfunction.30) However, those pro-inflammatory cytokines were detected in serum, thus, the sources of pro-inflammatory cytokines are unknown, because exercise induced skeletal muscle damage would also trigger inflammation, so it is incautious to conclude that cardiac dysfunction after IE is associated with increased expression of pro-inflammatory cytokines based on serum data. Our study histologically observed inflammatory cells infiltrated the RV of IE rats, which suggested that inflammation potentiates exercise induced right ventricular damage and fibrosis. Involvement of inflammation in exercise induced right ventricular damage and fibrosis were also supported by increased protein expression of MCP-1 and IL-1β in the RV following long-term IE. In animal studies, tumor necrosis factor (TNF) α has been detected in exercise induced atrial fibrosis, and atrial fibrosis was attenuated after TNFα inhibition.17) Our study is in line with other studies demonstrating inflammation is critical for the formation of exercise induced MF. Importantly, the TNFα dependent signaling pathway has been demonstrated to be essential for IE induced atrial fibrosis and atrial arrhythmia.17) The relationship between inflammation and fibrosis in myocardial infarction model was well reviewed by Prabhu and Frangogiannis.8) To summarize, myocardial infarction damage triggered an inflammatory response, this response cleared dead cells and matrix debris, thereafter inflammation was suppressed, and reparative cells were activated at the same time, then MF was formed. Although there may have some differences, in our study, similar right ventricular damage and fibrosis was detected after long-term IE. Therefore, inflammation is the potential pathological link between long-term IE induced right ventricular damage and fibrosis. If the underlying mechanisms of how MCP-1 and IL-1β mediate exercise induced right ventricular fibrosis are established, therapeutic modulation of the myocardial inflammatory response may hold promise for the prevention of long-term IE induced MF. Clearly, additional studies will be required to address the underlying mechanisms for MCP-1 and IL-1β mediated right ventricular damage and fibrosis with IE.
In conclusion, Long-term IE induced right ventricular damage is pathological, and the following inflammatory response is essential in the development of exercise induced MF.
Go to :
 XML Download
XML Download