

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Adult-onset Still's disease (AOSD) is a rare condition, with a prevalence ranging from 1 to 34 cases per million individuals worldwide [1]. The etiology of this autoimmune systemic inflammatory disease remains unknown. At present, no definite diagnostic radiological or laboratory results have been identified for AOSD [123].
The major symptoms of AOSD include spiking fever, rash, and arthritis [4]. The symptoms of AOSD include fever, which occurs in 60%–100% of cases, and arthralgia that mainly develops at the wrists, knees, and ankles in 70%–100% of cases. Moreover, a macular or maculopapular salmon-pink skin rash may appear mainly on the trunk or proximal limb, along with a fever spike. Sore throat is an early symptom of AOSD, and reportedly occurs in 69%–92% of cases [14]. Other potential manifestations include splenomegaly and lymphadenopathy, and reactive hemophagocytic lymphohistiocytosis. In addition, various other rare complications have been reported, including myocarditis, tamponade, and multiple organ failure [1456]. The other accompanying symptoms and signs include enlargement of the lymph nodes, myalgia, splenomegaly, hepatomegaly, pleurisy, pericarditis, and abdominal pain [1]. AOSD is primarily diagnosed based on clinical findings, and requires the exclusion of various other differential diagnoses, such as infectious conditions, neoplasm, and connective tissue disease [5].
AOSD occurs relatively frequently in people aged around 36 years. Its incidence is greater among women, and the second trimester of pregnancy and postpartum period are associated with an increased risk of recurrence [1]. We describe an 18-year-old young male patient who was diagnosed with AOSD based on the clinical features observed during rehabilitation treatment after the diagnosis of ischemic stroke.
CASE DESCRIPTION
An 18-year-old man with no any underlying disease was admitted to the emergency room at another hospital due to headache, nausea, and vomiting on 1 November 2015. No specific manifestations were noted on brain images or on physical examination, and the patient was discharged after his symptoms improved. However, he temporarily exhibited dysarthria, and ataxia and memory deterioration also developed on 27 November 2015. He was admitted to the emergency department on 30 November 2015.
Multiple embolic infarction was diagnosed by magnetic resonance imaging (MRI), and the patient received treatment at a hospital in his hometown. The patient exhibited worsening gait disturbance and cognitive impairment during this hospitalization. He was therefore transferred to our hospital on 15 December 2015.
The patient still complained gait disturbance, dizziness, dysarthria and memory impairment. Brain MRI (Philips, Achieva, Amsterdam, Netherlands) performed at our hospital indicated a manifestation of multifocal cerebral infarction on pons, middle cerebellar peduncle, cerebellum, internal capsule, corpus callosum, basal ganglias, corona radiata and cortical/subcortical area of both cerebral hemisphere (Fig. 1). There are no definite irregularities in intracranial vessels on MRI findings. Neck angio computed tomography (CT) finding was no definite abnormality. The antinuclear antibody (ANA), rheumatoid factor (RF), and anticardiolipin antibody IgG and IgM levels were normal. Tests for liver enzyme levels, human immunodeficiency virus (HIV), and syphilis also indicated normal results. No abnormal manifestation was observed in other blood tests or in a cerebrospinal fluid (CSF) study. The results of cardiac evaluation that including holter monitoring and echocardiogram tests were also normal. Considering the patient's age and clinical features, neurologists suspected that the multiple embolic infarction was likely due to vasculitis. Hence, daily 1,000 mg of methylprednisolone was injected for 5 days. After the regimen was switched to 60 mg of oral prednisolone, gradually and slowly tapering the medication during 3 months.
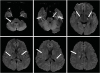 | Fig. 1Brain magnetic resonance imaging scan showing a multifocal diffusion restriction with a fluid-attenuated inversion-recovery signal change.
|
The patient's symptoms gradually improved after steroid treatment was initiated. He was transferred to the rehabilitation department of our hospital. At the time of this transfer, his Korean version of the Mini-Mental State Examination (K-MMSE) score was 15. Moreover, he was completely dependent on support for daily living activities.
After the comprehensive rehabilitation treatment, his K-MMSE score increased to 27 and his MBI score increased to 68. The patient was subsequently discharged at 2 months after the onset of stroke. He was readmitted to our hospital at 3 months after diagnosis for additional rehabilitation treatment and evaluation. Prednisolone was consistently reduced; he received 10 mg orally on alternate days.
The patient developed fever (38.9°C) at right after admission, while on prednisolone (10 mg) on alternate days. No evidence of infection was noted at that time. Hence, he was suspected to have developed fever as a result of long-term steroid administration. The prednisolone was therefore terminated after consultation with the neurology department. However, as the spike in fever beyond 39°C continued after prednisolone was stopped, every other day administration of 10 mg prednisolone was reinitiated. The aspartate transaminase (AST)/alanine aminotransferase (ALT) (IU/L) was 66/30 at 3 days after fever onset, indicating a manifestation of liver function test (LFT) elevation, and showed a consistent increase up to 74/123. The patient complained of lower back pain and a skin rash, knee arthralgia, sore throat also developed with LFT elevation (Fig. 2).
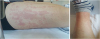
The allergic antibody tests of the patient were normal. Splenomegaly was confirmed based on abdominal CT (Siemens, Somatom definition AS+, Munchen, German) findings (Fig. 3), and blood tests indicated elevated white blood cells (15,800/μL; 91.6% neutrophils), erythrocyte sedimentation rate (7.0 mm/hr), and C-reactive protein (11.85 mg/dL). The results of stool and blood cultures, a throat swab were negative. The infection probability was assumed to be low due to the lack of infectious symptoms as consultation with the infectious disease department.
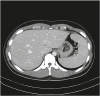 | Fig. 3Abdominal computed tomography scan showing splenomegaly, which is a characteristic finding of Still's disease.
|
An additional evaluation was performed to examine the possibility of autoimmune disease, and dilute Russell viper venom test was detected. The patient was referred to the rheumatology department. The patient was be ruled out infectious, neoplastic, and other auto-immune disorders. We confirmed that the patient had AOSD by Yamaguchi's diagnostic criteria. Symptoms and signs of the patient improved by a high dose administration of steroids at 2 weeks after the fever onset. The serum ferritin level was mild elevated at 355 μg/L (normal 20–300 μg/L) after steroid treatment. The patient exhibited recurring high fever, as well as arthralgia, typical rash, and leukocytosis, thus fulfilling all the major criteria of Yamaguchi's criteria. Among the minor criteria, sore throat and splenomegaly were identified on abdominal CT, along with elevated LFT parameters. The following day, high dose steroid therapy were initiated, and warfarin medication was additionally administered. The patient was treated with maintenance therapy with steroid at outpatient clinic.
DISCUSSION
AOSD is a systemic inflammatory illness that develops in young individuals. This disorder has a peak prevalence at 2 age ranges, 15–25 years and 36–46 years [27].
At present, only a few diagnostic criteria have been proposed. Among these, Yamaguchi's criteria are known to be the most sensitive [3]. According to Yamaguchi's classification, high fever (> 39°C), arthralgia for > 2 weeks, typical rash, and granulocytic leukocytosis (> 10,000, > 80% granulocytes) are major criteria. Sore throat, lymphadenopathy and/or splenomegaly, LFT abnormalities, and negative ANA and RF test results are minor criteria. AOSD is diagnosed in accordance with Yamaguchi's classification when at least 5 criteria are present, including 2 major criteria.
Stroke is a very rare complication in cases with AOSD [58]. Kurabayashi et al. [8] have reported cerebral hemorrhage complications in AOSD patients and considered vasculitis resulting from AOSD was the cause of this. There is also a case report and review for thrombotic microangiopathy that platelet microthrombi occlude small vessels in AOSD [9]. Yanai et al. [10] suggested association between AOSD and systemic vasculitis by presenting pathologic findings that compatible with leukocytoclastic vasculitis of salmon-pink rash and elevated markers for vasculitis in AOSD. Based on these studies, we assumed that the multiple cerebral infarction was related to the pathology of vasculitis in our present case.
Corticosteroids which was effective in this patient are currently used as the first-line treatment for this condition [1]. Aspirin or nonsteroidal anti-inflammatory drugs, disease modifying anti-rheumatic drugs, and methotrexate have been considered as treatment options for AOSD. Intravenous immunoglobulin, TNF-α blockers, interleukin (IL)-1β antagonists and IL-6 antagonists have also shown effect in AOSD [1256].
Although various pathological factors have been proposed for AOSD, including genetics, viral factors, and immunity, the main cause remains unknown. The probability of stroke incidence is very low in young individuals, and the identification of the underlying cause is vital for preventing its recurrence. Cerebral infarction of cardiac origin along with hematologic abnormalities are among the most common causes of stroke in younger people. But, other various causes have also been reported like nonatherosclerotic vascular disease [11]. Thus, if cerebral infarction is accompanied by unusual clinical features as non-infectious fever, rash, and arthralgia, AOSD should be considered when identifying the cause of stroke in young individuals. And it is important to be aware of the serial aspects of symptoms and signs.
 XML Download
XML Download