

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Although the nitric oxide (NO) plays a physiological role, which is necessary for vasodilation, its excess production induces several pathophysiological states.1 Excess production of NO can combine with superoxide anion radical, which causes cardiovascular disease, asthma, or arthritis.23 The inducible nitric oxide synthase (iNOS) plays a key role in the production of NO for the pathophysiological state. Recently, assays to search for the compounds with the inhibitory effect on NO formation are often used to test anti-inflammatory activity. The inhibitory effect on NO formation is frequently measured in lipopolysaccharide (LPS)-induced mouse macrophage cells. In the present study, this assay was performed in LPS-induced macrophage RAW 264.7 cells.
Caryopteris incana (Verbenaceae) is a perennial herb, which is used to treat cough, arthritis, and eczema in Oriental medicinal society.45 The constituents belonging to iridoid glycosides, phenylethanoid glycosides,56 and diterpenoids6 have been reported. In this study, the MeOH extract was divided into two fractions, the CHCl3 fraction and BuOH fraction, the former contains less polar substances and the latter does more polar ones. Since C. incana is a very flavoring plant, the essential oil was also prepared from this plant material to test an iNOS assay and to analyze the constituents.
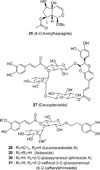
When the inhibition activity on iNOS were tested, the CHCl3 fraction and essential oil significantly inhibited the production of NO, but the BuOH fraction did not. Because the constituents of the essential oil will share those of the CHCl3 fraction, the essential oil was analyzed by gas chromatography (GC)-mass spectroscopy (MS). Furthermore, the six glycosides of an iridoid glycoside (8-O-acetylharpagide) and five phenylethaoid glycosides (caryopteroside, acteoside, phlinoside, 6-O-caffeoylphlinoside, leucosceptoside A) were quantitatively analyzed using an HPLC method.
Experimental
Instruments and reagents
Capillary gas chromatography (GC)-mass spectrometry (MS) used for analysis of the essential oil was an MSD 5975N (Agilent technologies, USA) equipped with HP6890 GC (Agilent technologies, USA) and HP-7683 auto-sampler (Agilent technologies, USA). GC column used for GC-MS analysis was 5MS (30 m × 0.25 mm i.d., 0.25 µm film thickness)
HPLC system consisting of two Prostar 210 pumps, a Prostar 325 UV-Vis detector, and a Shiseido Capcell PAK C18 column (5 µm, 4.6 mm × 250 mm) equipped with MetaTherm temperature controller were used for quantification. The HPLC grade solvent used for the mobile phase was purchased from J.T. Baker (Philisburg, NJ, USA). The stationary phase column chromatography used for isolation was silica gel 60 (70 – 230 mesh, Merck, Germany) and Diaion HP-20 (Mitsubishi Chemica. Co., Japan).
Plant material
The leaves of C. incana was collected on August in Wonju, Gangwon-do, Korea and then dried. The dried leaves were cut prior to extraction. This plant was identified by Prof. Byong-Min Song (Department of Forest Science, Sangji University). A voucher specimen (natchem-#92) was deposited in the Laboratory of Natural Products Sciences, Department of Pharmaceutical Engineering, Sangji University.
Steam distillation
To extract essential oil from the plant material, 300 g of the plant material and distilled for 3 h in the steam distillation apparatus. The distilled suspension was partitioned with diethyl ether three times, and further dehydrated with anhydrous sodium sulfate. Drying of the diethyl ether-soluble portion led to the preparation of essential oil (0.57 g, 0.58 mL). Density of this essential oil was calculated to be 0.982 g/mL.
Measurement of GC-MS
GC chromatography was performed at 250 ℃ of the injection port temperature with a splitless mode (injection mode) and 1 µL of injection volume. Helium as the carrier gas was flowed at 1.0 mL/min. Column temperature was increased as the programmed method. Initial temperature was maintained at 60 ℃ for 1.0 min. This temperature was increased to 130 ℃ at the rate of 20 ℃/min, then to 190 ℃ at 5 ℃/min, and finally increased to 240 ℃ at 20 ℃/min. The final temperature was maintained for 1 min. In MS, the 70 eV was used for ionization of molecules as the electron impact mode. Transfer line temperature was 280 ℃ and solvent delay time was 2 min. Measurement of mass spectra was performed with scan mode, where acquisition mode was m/z 40 – 500. Identification of molecules was performed by matching each spectrum with that of internal library (Wiley W9NO8 program) and confirmed at more than 90% quality ratio.
The number (1 – 15) of compounds of which structures were identified were given.
Extraction and fractionation
The plant material of the leaves of C. incana (440 g) was extracted with MeOH under reflux for 5 h three times. The extracted solution was filtered and evaporated on a rotatory evaporator under reduced pressure. The concentrated extract was further freeze-dried to give a MeOH extract (130.0 g). The MeOH extract (128 g) was suspended in H2O and then partitioned with CHCl3 three times. A CHCl3-soluble portion was concentrated to dryness to give a CHCl3 fraction (19 g). A residual aqueous layer was partitioned with BuOH three times. A BuOH-soluble portion was concentrated in vacuo and then freeze-dried to give a BuOH fraction (48.1 g). The aqueous layer was dried in vacuo and subjected to dreeze-drying to give a H2O fraction (67.3 g).
After the solvent-fractionation, successive column fractionation was performed using a Diaion HP-20 column. The BuOH fraction (40.3 g) was subjected to Diaion HP-20 column chromatography. This column was eluted with 1.0 L to remove sugars or ionic substances. Successive elution was conducted with each 1.0 L solvent of 25% MeOH, 50% MeOH, 75% MeOH, and 100% MeOH, every eluted solution was concentrated to produce a 25% MeOH extract (5.3 g), 50% MeOH extract (1.9 g), 75% MeOH extract (8.3 g), and 100% MeOH (6.2 g) extract.
Cell culture
Mouse macrophage RAW 264.7 cells, obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA), were cultured in DMEM supplemented with 10% heat-inactivated fetal bovine serum (FBS) and antibiotics-antimycotics (PSF; 100 units/ml penicillin G sodium, 100 ng/mL streptomycin, and 250 ng/mL amphotericin B). The cells were maintained at 37 ℃ under a humidified atmosphere containing 5% CO2.
Cell viability assay
MTT solution (final concentration of 500 µg/mL) was added to each well and incubated for 4 h at 37 ℃. The culture media was removed, and dimethyl sulfoxide (DMSO) was added. The absorbance was measured at 570 nm, and percent survival was determined by comparison with a control group (LPS+).
iNOS assay
RAW 264.7 cells were cultured in 10% FBS–DMEM, and seeded in 24-well plates (2 × 105 cells/mL). The next day, culture media was changed to 1% FBS-DMEM and samples were treated. After 1 h, 1 µg/mL of LPS was added, except LPS- control, to stimulate NO production. Cells were incubated for an additional 18 h and the amount of NO production in culture media was determined by Griess reaction. Briefly, 180 µL of Griess reagent (0.1% N-(1-naphthyl) ethylenediamine dihydrochloride in H2O and 1% sulfanilamide in 5% H3PO4) was added to 100 µL of culture media. Then absorbance was measured at 540 nm, and nitrite concentration was determined by comparison with a sodium nitrite standard curve. Percent inhibition was calculated using following formula: [1 − (NO level of test samples/NO levels of vehicle-treated control)] × 100. The IC50 value, the sample concentration resulting in 50% inhibition of NO production, was calculated through non-linear regression analysis using Table Curve 2D v5.01 (Systat Software Inc., San Jose, CA, USA).
Isolation
The 75% fraction (10.0 g) was chromatographed over silica gel column (SiO2 180 g, Φ 4.5 cm × 30 cm) using the eluent CHCl3-MeOH-H2O (65 : 35 : 10, lower phase) and collected by each 50 ml to give 35 aliquots (#1 – #35). These 35 aliquots were checked on TLC visualized by UV light and 50% H2SO4, and then combined into five groups. The six groups named CI-1 (#6 – #8), CI-2 (#12 – #14), CI-3 (#15 – #16), CI-4 (#21 – #23), CI-5 (#26 – #29), and CI-6 (#29 – #35) were concentrated to afford compounds 26 – 31, respectively.
Compound 26 (8-O-acetylharpagide)
UV λmax (MeOH) nm (log ε): 203 (3.83); IR νmax (KBr) cm−1: 3357 (broad, O-H), 2914 (C-H), 1712 (C=O), 1653 (C=C), 1375 (CH3), 1237 (C-O), 1117, 1077 (Glycoside C-O); 1H-NMR (600 MHz, CD3OD) and 13C-NMR (150 MHz, CD3OD) δ: literature.5
Compound 27 (caryopteroside)
UV λmax (MeOH) nm (log ε): 268 (4.06), 329 (4.14); IR νmax (KBr) cm−1: 3403 (broad, O-H), 2940, 2883 (C-H), 1724, 1708, 1687 (C=O), 1604, 1517 (aromatic C=C), 1454, 1369, 1273 (phenolic C-O), 1175 (tertiary C-O), 1074 (secondary C-O), 1034 (glycoside C-O), 849, 815; 1H-NMR (600 MHz, CD3OD) and 13C-NMR (150 MHz, CD3OD) δ: literature.4
Compound 28 (acteoside)
UV λmax (MeOH) nm (log ε): 328 (4.11); IR νmax (KBr) cm−1: 3403 (broad, OH), 2931, 2886 (C-H), 1698 (C=O), 1631 (olefinic C=C), 1604, 1516 (aromatic C=C), 1445, 1366, 1280 (phenolic C-O), 1161 (tertiary C-O), 1071 (secondary C-O), 1037 (glycoside C-O), 814; 1H-NMR (600 MHz, CD3OD) and 13C-NMR (150 MHz, CD3OD) δ: literature.10
Compound 29 (leucosceptoside A)
UV λmax (MeOH) nm (log ε): 313 (4.28); IR νmax (KBr) cm−1: 3392 (broad, OH), 2931 (CH), 1705 (C=O), 1634 (olefinic C=C), 1604, 1471 (aromatic C=C), 1373 (CH3), 1365, 1235 (phenolic C-O), 1169 (tertiary C-O), 1135 (secondary C-O), 1073 (glycoside C-O), 834; 1H-NMR (600MHz, CD3OD) and 13C-NMR (150MHz, CD3OD) δ: literature.11
Compound 30 (phlinoside A)
UV λmax (MeOH) nm (log ε): 323 (4.21); IR νmax (KBr) cm−1: 3404 (broad, O-H), 2926, 2885 (C-H), 1699 (C=O), 1631 (olefinic C=C), 1604, 1517 (aromatic C=C), 1445, 1366, 1274 (phenolic C-O), 1167 (tertiary C-O), 1073 (secondary C-O), 1039 (glycoside C-O), 815; 1H-NMR (600 MHz, CD3OD) and 13C-NMR (150 MHz, CD3OD) δ: literature.10
Compound 31 (6-O-caffeoylphlinoside)
UV λmax (MeOH) nm (log ε): 332 (3.98); IR νmax (KBr) cm−1: 3384 (broad, O-H), 2934, 2891 (C-H), 1698 (C=O), 1631 (olefinic C=C), 1604, 1519 (aromatic C=C), 1445, 1366, 1281 (phenolic C-O), 1162 (tertiary C-O), 1115 (secondary C-O), 1070 (primary C-O), 1039 (glycoside C-O), 815; 1H-NMR (600 MHz, CD3OD) and 13C-NMR (150 MHz, CD3OD) δ: literature.5
Preparation of standard- and sample solutions
The stock solution (1000 µg/mL) was prepared by dissolving each compound in MeOH and preserved under the temperature of 4 ℃. To prepare working standard solutions, the stock solution was serially diluted. Regression equations were determined by calculation of the peak area (y axis) at six concentrations (x axis, µg/ml). Sample solutions were prepared by dissolving the MeOH extract, BuOH fraction, the 25%, 50%, 75%, and 100% MeOH fraction in MeOH. These solutions were filtered through disposable syringe filter unit (0.50 µm, Sismic-25JP Advantec, Japan) prior to injection.
HPLC analytical method
Two solvents, solvent A (H2O with 0.05% acetic acid, v/v) and solvent B (MeOH) were used in this method. The linear gradient elution of the solvents was programmed as follows: 0 – 20 min (15 → 60% B), 20 – 21 min (60 → 100% B), 21 – 25 min (100% B), 25 – 27 min (100 → 15% B), and 27 – 30 min (15% B). The flow rate and column temperature was set constantly at 1.0 mL/min and 40 ℃, respectively. The detection wavelength was fixed at 205 nm. The analytical method was validated in terms of linearity and sensitivity, referring to the guidance of international conference on harmonization (ICH). The R2 value was used to assess the linearity of regression equation. LOD (limit-of-detection) and LOQ (limit-of-quantification) was used as the measure of sensitivity, where LOD and LOQ were determined at signal-to ratios (S/N ratio) of 3 and 10, respectively.
Result and Discussion
As shown in Table 1, the CHCl3 fraction and the essential oil greatly inhibited iNOS activities in LPS-activated macrophage RAW 264.7 cells, demonstrating that the two samples have anti-inflammatory activities. However, the BuOH fraction and its five compounds including an iridoid (8-O-acetylharpagide)- and five phenylethanoid glycosides (caryopteroside, acteoside, phlinoside, 6-O-caffeoylphlinoside, and leucosceptoside A) had no activity (data not shown). The IC50s on the iNOS activity were 16.4 (CHCl3 fraction) and 23.8 µg/mL (essential oil), respectively. Furthermore, the CHCl3 fraction was cytotoxic at the 80 µg/mL concentration while the essential oil was not, indicating that the essential oil may be more advantageous for the anti-inflammatory activity. Since the essential oil shares volatile substances with the CHCl3 fraction containing less polar substances, this oil was analyzed by GC-MS. Twenty-five components were identified by matching each mass spectrum with the data of MS library. Of compounds 1 – 14, twelve peaks besides compounds 1 and 2 belonged to monoterpenes. From the peak 15 (α-copaene) at 10.31 min (tR) to 25 (α-bisabolene epoxide) at 13.85 min on the GC chromatogram (Fig. 2), eleven compounds were sesquiterpenes. Considering the peak area, the amount of sesquiterpenes was relatively low compared to those of monoterpenes.
Most compounds were cyclic terpenes except for the two simple alcohols (3-hexen-1-ol, 1-octen-3-ol) and the two acylic monoterpens (linalool and cis-citral). Cyclic monoterpenes were shown including 1-trans-pinocarveol, cis-citral, β-phellandren-8-ol, 2(10),-pinen-3-one, terpendiol I, myrtenol, 1-verbenone, trans-(+)-carveol, terpendiol II, 1,3,8-p-menthatriene, and (+)-myrtenyl acetate. The identification was confirmed by matching each spectrum with the database of the Wiley 10th version library. In Table 2, the molecular ion peak, base peak (100% relative intensity) and other significant peaks were shown. Of the 25 peaks, three major components were trans-pinocarveol (4, 18.78%), cis-citral (5, 13.53%), pinocarvone (7, 18.37%) (Fig. 1), where the percentage of peak area was shown in the parenthesis. Although a number of peaks of essential oils are shown in the chromatogram, a few compounds usually occupies predominant peak area of the compounds. Therefore, the essential oil of C. incana could be used based on the high percentage of trans-pinocarveol (4), cis-citral (5), and pinocarvone (7). Regarding sesquiterpenes, the eleven compounds including α-copaene, γ-cadinene, γ-2-cadinene, δ-cadinene, β-copaene-4α-ol, α-campholenal, ledol, α-cedrene, τ-muurodol, caryophyllene epoxide, and α-bisabolene epoxide were identified. The 25 compounds identified in this study represented 85.5% quantity of the total essential oil.
The BuOH fraction and its isolates of the glycosides did not inhibit NO formation in LPS-activated macrophage 264.7 cells. Since these glycosides could be active in other assays through the possible hydrolysis in the living system, the quantitative analysis of the phenolic glycosides were attempted using an HPLC method. The structure of six compounds were 8-O-acetylharpagide,5 caryopteroside,4 acteoside,10 phlinoside,5 6-O-caffeoylphlinoside,5 and leucosceptoside A11 were identified by comparisons of 1H- and 13C-NMR spectroscopic data with literatures. One is an iridoid glycoside, and the other five are phenylethanoid glycosides, as shown in Fig. 3. Diaion HP-20 is the stationary phase with a structure of a spherical porous particle consisting of polystyrene polymers. In the present study, the HPLC analysis on six glycosides was performed in the MeOH extract, CHCl3- and BuOH fractions, and each fractions eluted with MeOH-H2O solvents over a Diaion HP-20 column (Fig. 4). Retention times of six glycosides were shown in Table 3. The wavelength of 205 nm was chosen because only 8-O-acetylharpagide is not sensitive at 280 nm and 360 nm. Each regression equation was established after measuring the peak area at every six concentration (n = 4). The R2 values more than 0.999 in the range of 31.25 – 1000.0 µg/mL established the linearity of the six equations. Every value of LOD and LOQ was sufficiently low indicating that they are very sensitive for the detection and quantification. As shown in Table 4, the content of six compounds were determined in the MeOH extract as the following order: caryopteroside (162.35 mg/g) > 8-O-acetylharpagide (93.28mg/g) > 6-O-caffeoylphlinoside (28.15 mg/g) > phlinoside A (22.60 mg/g) > leucosceptoside A (16.87 mg) > acteoside (7.05 mg/g). These substances were quantitatively higher in the BuOH fraction than in the CHCl3 fraction. In particular, 8-O-acetylharpagide was very rich (86.4%), but other five substances were not detected in the 25% MeOH fraction. In addition, caryopteroside occupied 11.2% of the 75% MeOH fraction. However, the quantity of acteoside and phlinoside was relatively low. In spite of no inhibition of the six glycosides on NO formation, other biological activities of them have been reported. Therefore, these analytical data will be useful for further research of C. incana.
In conclusion, the essential oil obtained from C. incana inhibited NO formation in macrophage 264.7 cells. GC-MS analysis of the essential oil led to the identification of 25 volatile substances and the finding of three main substances, trans-pinocarveol (4, 18.78%), cis-citral (5, 13.53%), and pinocarvone (7, 18.37%). Furthermore, the high content of caryopteroside (162.35 mg/g), 8-O-acetylharpagide (93.28 mg/g), and 6-O-caffeoylphlinoside (28.15 mg/g) were also elucidated in this plant by HPLC.

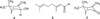





 XML Download
XML Download