

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Pfeiffer syndrome is a rare genetic disorder characterized by premature fusion of certain skull bones (craniosynostosis) and anomalies of the face and extremities. Additional anomalies in Pfeiffer syndrome may include low-set ears, external auditory canal stenosis or atresia, aqueductal stenosis, hydrocephalus, cerebellar and brain stem herniation, hydronephrosis, pelvic kidney, hypoplastic gallbladder, and visceral malformations.1 Since this disorder was first reported by Pfeiffer in 1964, new findings have been described. However, accompanying vertebral malformations are rare.2 Here we report a case of Pfeiffer syndrome with coccygeal anomaly suspected to be clinically Pfeiffer syndrome type 2. Molecular genetic testing revealed fibroblast growth factor receptor (FGFR) 2 gene mutation, thus confirming the diagnosis.
Case
A female infant was born at 37+1 weeks of gestation via normal spontaneous vaginal delivery. Her mother was referred to our hospital at gestation of 34 weeks because of suspected multiple anomalies of fetus. A detailed fetal sonographic examination was performed in our hospital and the fetus was confirmed to have an abnormal head shape like a cloverleaf and coccygeal anomaly. Her mother was 29 years old. During pregnancy, she was taking levothyroxine to treat her hypothyroidism. Her parents were both phenotypically normal Koreans who did not have any history of consanguinity.
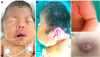
The patient's Apgar scores at 1 and 5 minutes were 9 and 10, respectively. Birth weight was 3,200 g (50–90 percentile), length 48.6 cm (50–90 percentile), and head circumference was 32 cm (10–50 percentile). Physical examination showed cloverleaf-shaped head, proptosis, hypertelorism, low-set ear, maxillary hypoplasia, mandibular prognathism, cleft palate, and bilateral elbow ankylosis making both arms bend into the radial side with limited extension of elbows less than 90 degrees. Neither leg showed any specific joint stiffness or limitation of movement. The overlying skin defect in the coccygeal region raised suspicion of a spinal anomaly and protrusion of underlying structure that made it look like a human tail (Fig. 1).
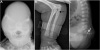
Plain radiographs of the skull showed bulging of both temporal fossae and fusion of sagittal and coronal sutures, producing a cloverleaf skull. Plain radiographs of her elbows showed fusion of humeroulnar, humeroradial, and radioulnar joints. A lateral spine view showed straightened spinal curve with the sacrococcygeal bone abruptly directed posteriorly (Fig. 2).
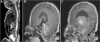
Sacrococcygeal eversion was more evident on spine magnetic resonance imaging. There was hydrocephalus in the 3rd ventricle, tonsillar herniation, and crowd foramen magnum on brain magnetic resonance imaging (Fig. 3). There was no sign of tethered cord, meningocele, or myelomeningocele associated with the coccygeal skin defect.
The patient did not require respiratory support. Her vital sign was stable during hospital days. She tolerated feeds with tube and Habermann feeder well. She achieved full enteral feeding on the 6th day of life. Her basic laboratory tests were normal. Her thyroid function test was also normal. Ultrasonography of the abdomen revealed malrotation of intestines with superior mesenteric artery and vein reversal. Echocardiography showed small atrial septal defect, multiple small calcifications in chordae of mitral and tricuspid valves, and mild noncompacted myocardium of left ventricle. Auditory brainstem response threshold test confirmed that she had hearing disorders required hearing rehabilitation and/or surgery. She did not have any abnormal finding on her ophthalmologic examination other than proptosis.
After genomic deoxyribonucleic acid was isolated from peripheral blood, polymerase chain reaction was performed to analyze mutation of FGFR 2 gene for exons 7 and 8. Results revealed c. 870G>C (p.Trp 290 Cys) mutation (Fig. 4).
On the 18th day of life, the 4th coccyx at her skin defect site was removed. Follow-up sonography of brain revealed aggravation of hydrocephalus. Because she needed correction surgeries to release the prematurely fused skull and promote normal brain and skull growth, the baby was transferred to another hospital on the 69th day of life.
Discussion
Originally described in a German family in 1964, Pfeiffer syndrome is one of the most common craniosynostosis syndromes (particularly of coronal sutures and sometimes sagittal sutures). The inheritance pattern of Pfeiffer syndrome is usually autosomal dominant but sometimes sporadic.34 The frequency of its occurrence is about 1 in 100,000 live births.3 It is rarer in Asian population, with only a few cases reported in Korea.567 Characteristic features of this syndrome include craniosynostosis, broad thumbs, and broad great toes. Other anomalies and internal organ malformations may be occasionally associated with other craniosynostosis syndromes such as Crouzon syndrome8 and Apert syndrome.3
This syndrome is usually noted in the newborn period or later. Although prenatal diagnosis is difficult and mainly based on the presence of a cloverleaf-shaped skull deformity,3 a careful three-dimensional ultrasound examination can lead to early prenatal diagnosis even for cases without cloverleaf-shaped skull.9 The syndrome has three clinical subtypes10 with various clinical features and prognostic implications.1 Patient with type 1 have mild phenotype with brachycephaly, mid-facial hypoplasia, finger and toe abnormalities, and near-normal intelligence. Type 2 shows a cloverleaf-shaped head, severe proptosis, and central nervous system involvement caused by more extensive fusion of skull bones, with potential elbow ankylosis or synostosis. Type 3 is similar to type 2. However, type 3 does not have cloverleaf-shaped skull. Types 2 and 3 are more severe than type 1 with poorer neurodevelopmental outcomes. Considerable clinical overlap may occur among the three subtypes. Prognostic implications vary according to types. Patients with type 1 generally have good outcomes. It can be dominantly inherited in some families. Both types 2 and 3 are sporadic with poor neurodevelopmental outcomes and early death. The present case had ocular proptosis and typical cloverleaf-shaped skull. Hence, type 2 was considered.
After birth, the prognosis of Pfeiffer syndrome depends on associated anomalies. A series of staged surgeries are needed to release prematurely closed sutures.9,11 Early treatment may prevent secondary complications such as hydrocephaly and cognitive impairment. Treatments involves a multidisciplinary approach with ultimate surgical fixation of the cranial deformity to prevent further sequelae.
Molecular genetic testing is important to confirm the diagnosis of Pfeiffer syndrome. Mutations in FGFR 1, 2, or 3 can affect craniofacial and skeletal development.12 Pfeiffer syndrome can be caused by heterozygous mutations in FGFR 1 gene2 on chromosome 8 or in FGFR 2 gene3 on chromosome813 Rossi et al.14 have reported common FGFR 1 P252R mutation in four affected families, all of which demonstrated characteristic malformation of feet with variable or absent skull involvement.
Tartaglia et al.15 have reported a de novo G-to-C transversion in exon IIIa of the FGFR 2 gene, resulting in a Trp-to-Cys missense mutation at codon 290 (T290C; 176943.0019). The patient had cloverleaf-shaped skull deformity as well as other typical ocular, hand, and foot anomalies without coccygeal anomaly seen in Pfeiffer syndrome. Schaefer et al.16 have also found a T290C mutation in a case of Pfeiffer syndrome type 2. The infant had cloverleaf-shaped skull, proptosis, radioulnar synostosis, and broad thumbs, and great toes.
In rare cases, coccygeal anomaly may be accompanied by Pfeiffer syndrome, which could be confirmed by genetic testing. Gonzales et al.17 have reported three fetuses diagnosed prenatally with severe Pfeiffer syndrome. All of them had the same heterozygous mutation in the FGFR 2 gene. All three patients had vertebral anomalies, including cervical, thoracic, and lumbar fusion. Sacrococcygeal eversion was also present in two Korean cases of Pfeiffer syndrome.25 True human tail usually occurs in the lumbosacral region. It can move and contract. On the contrary, pseudo-tail is anomalous prolongation of the coccygeal vertebrae that resembles true tail.18 In our case, the dermal appendage and eversion of vertebrae in the coccygeal region resembled human tail.
In summary, we observed associations of sporadic FGFR 2 mutation and coccygeal anomaly with Pfeiffer syndrome type 2. Molecular genetic testing should be considered for any type of Pfeiffer syndrome to obtain definite diagnosis.

 XML Download
XML Download