

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Abstract
Monogenic autoimmune diseases (AD) present as lupus-like clinical manifestations with recurrent fever or various vasculopathies. Recurrent fever with an elevation of acute phase reactants and various skin lesions are similar in monogenic AD and autoinflammatory disease. The molecular pathogenesis of adult systemic erythematosus can be understood through monogenic AD based on gene defects: complement, apoptosis, interferonopathy via nucleic acid sensing, tolerance, raso-pathies, and others. Skin vasculopathy with chilblains and livedo reticularis, interstitial lung disease, and panniculitis are common occurrences in type I interferonopathy. Some syndromes have been reported to present with autoimmune inflammation and the general clinical findings, including cerebral calcification. Various clinical manifestations in monogenic AD present in accordance with the gene loss- or gain-of-function mutations involved. The monogenic AD for the early onset of more severe lupus-like symptoms or vasculopathy needs to be considered. Furthermore, clinical trials were conducted via targeted therapy for related molecular pathways, because conventional treatments were not effective in managing monogenic AD.
Go to : 
REFERENCES
1. Weiss JE. Pediatric systemic lupus erythematosus: more than a positive antinuclear antibody. Pediatr Rev. 2012; 33:62–73. quiz 74.

2. Petri M, Orbai AM, Alarcon GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and validation of the systemic lupus international collaborating clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012; 64:2677–86.
3. Tsokos GC. Systemic lupus erythematosus. N Engl J Med. 2011; 365:2110–21.
4. Qing X, Putterman C. Gene expression profiling in the study of the pathogenesis of systemic lupus erythematosus. Autoimmun Rev. 2004; 3:505–9.
5. Rekvig OP, Van der Vlag J. The pathogenesis and diagnosis of systemic lupus erythematosus: still not resolved. Semin Immunopathol. 2014; 36:301–11.
6. Zen M, Gatto M, Domeneghetti M, Palma L, Borella E, Iaccarino L, et al. Clinical guidelines and definitions of autoinflammatory diseases: contrasts and comparisons with au-toimmunity-a comprehensive review. Clin Rev Allergy Immunol. 2013; 45:227–35.
7. Doria A, Zen M, Bettio S, Gatto M, Bassi N, Nalotto L, et al. Autoinflammation and autoimmunity: bridging the divide. Autoimmun Rev. 2012; 12:22–30.
8. Moghaddas F, Masters SL. Monogenic autoinflammatory diseases: Cytokinopathies. Cytokine. 2015; 74:237–46.
9. Costa-Reis P, Sullivan KE. Monogenic lupus: it's all new! Curr Opin Immunol. 2017; 49:87–95.
10. Lo MS. Monogenic lupus. Curr Rheumatol Rep. 2016; 18:71.
11. de Jesus AA, Goldbach-Mansky R. Newly recognized mendelian disorders with rheumatic manifestations. Curr Opin Rheumatol. 2015; 27:511–9.
12. Crow YJ. Type I interferonopathies: a novel set of inborn errors of immunity. Ann N Y Acad Sci. 2011; 1238; 91–8.
13. Kim H, Sanchez GA, Goldbach-Mansky R. Insights from mendelian interferonopathies: comparison of CANDLE, SAVI with AGS, monogenic lupus. J Mol Med (Berl). 2016; 94:1111–27.
14. Volpi S, Picco P, Caorsi R, Candotti F, Gattorno M. Type I interferonopathies in pediatric rheumatology. Pediatr Rheumatol Online J. 2016; 14:35.
15. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. 2014; 371:507–18.
16. Hiraki LT, Silverman ED. Genomics of systemic lupus erythematosus: insights gained by studying monogenic young-onset systemic lupus erythematosus. Rheum Dis Clin North Am. 2017; 43:415–34.
17. Vece TJ, Watkin LB, Nicholas S, Canter D, Braun MC, Guillerman RP, et al. Copa syndrome: a novel autosomal dominant immune dysregulatory disease. J Clin Immunol. 2016; 36:377–87.
18. Weill O, Decramer S, Malcus C, Kassai B, Rouvet I, Ginhoux T, et al. Familial and syndromic lupus share the same phenotype as other early-onset forms of lupus. Joint Bone Spine. 2017; 84:589–93.
19. Marlow AA, Peabody HD Jr, Nickel WR. Familial occurrence of systemic lupus erthematosus. JAMA. 1960; 173:1641–3.
21. Lintner KE, Wu YL, Yang Y, Spencer CH, Hauptmann G, Hebert LA, et al. Early components of the complement classical activation pathway in human systemic autoimmune diseases. Front Immunol. 2016; 7:36.
22. Miller EC, Atkinson JP. Overcoming C2 deficiency. Clin Immunol. 2012; 144:269–71.
23. Hauck F, Lee-kirsch MA, Aust D, Roesler J, Pessler F. Complement C2 deficiency disarranging innate and adaptive humoral immune responses in a pediatric patient: treatment with rituximab. Arthritis Care Res (Hoboken). 2011; 63:454–9.
24. Rieux-Laucat F, Le Deist F, Hivroz C, Roberts IA, Debatin KM, Fischer A, et al. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science. 1995; 268:1347–9.
25. Wu J, Wilson J, He J, Xiang L, Schur PH, Mountz JD. Fas ligand mutation in a patient with systemic lupus erythematosus and lymphoproliferative disease. J Clin Invest. 1996; 98:1107–13.
26. Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Moroy T. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet. 2000; 25:177–81.
27. Koyama R, Arai T, Kijima M, Sato S, Miura S, Yuasa M, et al. DNase gamma, DNase I and caspase-activated DNase cooperate to degrade dead cells. Genes Cells. 2016; 21:1150–63.
28. Rodero MP, Crow YJ. Type I interferon-mediated monogenic autoinflammation: The type I interferonopathies, a conceptual overview. J Exp Med. 2016; 213:2527–38.
29. Salzer E, Santos-Valente E, Keller B, Warnatz K, Boztug K. Protein kinase C δ: a gatekeeper of immune homeostasis. J Clin Immunol. 2016; 36:631–40.
30. Limnander A, Zikherman J, Lau T, Leitges M, Weiss A, Roose JP. Protein kinase C δ promotes transitional B cell-negative selection and limits proximal B cell receptor signaling to enforce tolerance. Mol Cell Biol. 2014; 34:1474–85.
31. Belot A, Kasher PR, Trotter EW, Foray AP, Debaud AL, Rice GI, et al. Protein kinase cdelta deficiency causes mendelian systemic lupus erythematosus with B cell-defective apoptosis and hyperproliferation. Arthritis Rheum. 2013; 65:2161–71.
32. Notarangelo LD, Kim MS, Walter JE, Lee YN. Human RAG mutations: biochemistry and clinical implications. Nat Rev Immunol. 2016; 16:234–46.
33. Walter JE, Lo MS, Kis-Toth K, Tirosh I, Frugoni F, Lee YN, et al. Impaired receptor editing and heterozygous RAG2 mutation in a patient with systemic lupus erythematosus and erosive arthritis. J Allergy Clin Immunol. 2015; 135:272–3.
34. Mor A, Philips MR, Pillinger MH. The role of Ras signaling in lupus T lymphocytes: biology and pathogenesis. Clin Immunol. 2007; 125:215–23.
35. Ragotte RJ, Dhanrajani A, Pleydell-Pearce J, Del Bel KL, Tarailo-Graovac M, van Karnebeek C, et al. The importance of considering monogenic causes of autoimmunity: a somatic mutation in KRAS causing pediatric Rosai-Dorfman syndrome and systemic lupus erythematosus. Clin Immunol. 2017; 175:143–6.
36. Tolmie JL, Shillito P, Hughes-Benzie R, Stephenson JB. The Aicardi-Goutieres syndrome (familial, early onset encephalopathy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis). J Med Genet. 1995; 32:881–4.
37. Ramantani G, Kohlhase J, Hertzberg C, Innes AM, Engel K, Hunger S, et al. Expanding the phenotypic spectrum of lupus erythematosus in Aicardi-Goutieres syndrome. Arthritis Rheum. 2010; 62:1469–77.
38. Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A. 2015; 167A:296–312.
39. Rice GI, Forte GM, Szynkiewicz M, Chase DS, Aeby A, Abdel-Hamid MS, et al. Assessment of interferon-related biomarkers in Aicardi-Goutieres syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study. Lancet Neurol. 2013; 12:1159–69.
40. de Vries B, Steup-Beekman GM, Haan J, Bollen EL, Luyendijk J, Frants RR, et al. TREX1 gene variant in neuropsychiatric systemic lupus erythematosus. Ann Rheum Dis. 2010; 69:1886–7.
41. Buers I, Nitschke Y, Rutsch F. Novel interferonopathies associated with mutations in RIG-I like receptors. Cytokine Growth Factor Rev. 2016; 29:101–7.
42. Hacohen Y, Zuberi S, Vincent A, Crow YJ, Cordeiro N. Neuromyelitis optica in a child with Aicardi-Goutieres syndrome. Neurology. 2015; 85:381–3.
43. Ravenscroft JC, Suri M, Rice GI, Szynkiewicz M, Crow YJ. Autosomal dominant inheritance of a heterozygous mutation in SAMHD1 causing familial chilblain lupus. Am J Med Genet A. 2011; 155A:235–7.
44. Gunther C, Kind B, Reijns MA, Berndt N, Martinez-Bueno M, Wolf C, et al. Defective removal of ribonucleotides from DNA promotes systemic autoimmunity. J Clin Invest. 2015; 125:413–24.
45. Lee-Kirsch MA, Gong M, Schulz H, Ruschendorf F, Stein A, Pfeiffer C, et al. Familial chilblain lupus, a monogenic form of cutaneous lupus erythematosus, maps to chromosome 3p. Am J Hum Genet. 2006; 79:731–7.
46. Richards A, van den Maagdenberg AM, Jen JC, Kavanagh D, Bertram P, Spitzer D, et al. C-terminal truncations in human 3′-5′ DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat Genet. 2007; 39:1068–70.
47. Al-Mayouf SM, Sunker A, Abdwani R, Abrawi SA, Almurshedi F, Alhashmi N, et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet. 2011; 43:1186–8.
48. Ozcakar ZB, Foster J 2nd, Diaz-Horta O, Kasapcopur O, Fan YS, Yalcinkaya F, et al. DNASE1L3 mutations in hypo-complementemic urticarial vasculitis syndrome. Arthritis Rheum. 2013; 65:2183–9.
49. Jain A, Misra DP, Sharma A, Wakhlu A, Agarwal V, Negi VS. Vasculitis and vasculitis-like manifestations in monogenic autoinflammatory syndromes. Rheumatol Int. 2018; 38:13–24.
50. Agarwal AK, Xing C, DeMartino GN, Mizrachi D, Hernandez MD, Sousa AB, et al. PSMB8 encoding the beta5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome. Am J Hum Genet. 2010; 87:866–72.
51. Arima K, Kinoshita A, Mishima H, Kanazawa N, Kaneko T, Mizushima T, et al. Proteasome assembly defect due to a proteasome subunit beta type 8 (PSMB8) mutation causes the autoinflammatory disorder, Nakajo-Nishimura syndrome. Proc Natl Acad Sci U S A. 2011; 108:14914–9.
52. Liu Y, Ramot Y, Torrelo A, Paller AS, Si N, Babay S, et al. Mutations in proteasome subunit beta type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum. 2012; 64:895–907.
53. Brehm A, Liu Y, Sheikh A, Marrero B, Omoyinmi E, Zhou Q, et al. Additive loss-of-function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production. J Clin Invest. 2015; 125:4196–211.
54. Kim H, Brooks KM, Tang CC, Wakim P, Blake M, Brooks SR, et al. Pharmacokinetics, Pharmacodynamics, and Proposed Dosing of the Oral JAK1 and JAK2 Inhibitor Baricitinib in Pediatric and Young Adult CANDLE and SAVI Patients. Clin Pharmacol Ther. 2018; 104:364–73.
55. Merrill JT, Wallace DJ, Petri M, Kirou KA, Yao Y, White WI, et al. Safety profile and clinical activity of sifalimumab, a fully human anti-interferon alpha monoclonal antibody, in systemic lupus erythematosus: a phase I, multicentre, dou-ble-blind randomised study. Ann Rheum Dis. 2011; 70:1905–13.
56. Petri M, Wallace DJ, Spindler A, Chindalore V, Kalunian K, Mysler E, et al. Sifalimumab, a human anti-interferon-alpha monoclonal antibody, in systemic lupus erythematosus: a phase I randomized, controlled, dose-escalation study. Arthritis Rheum. 2013; 65:1011–21.
Go to :
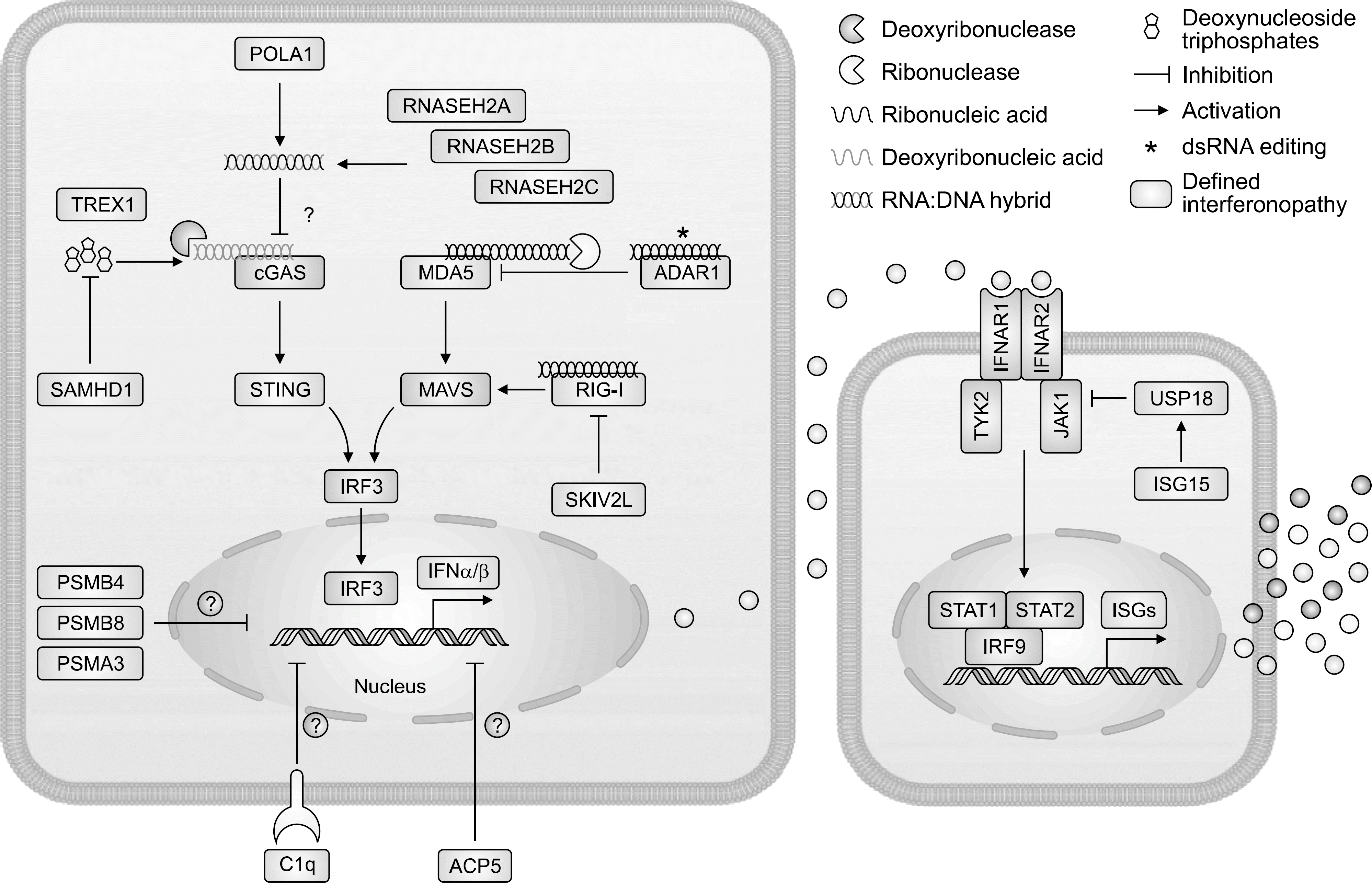 | Figure 1.Type 1 interferon signaling and type 1 interferonopathies as currently assigned. This figure is adapted from Rodero et al. J Exp Med 2016;213:2527-38 [28]. ADAR1: adenosine deaminase RNA specific 1, ACP5: acid phosphatase 5, C1q: complement 1q, cGAS: GMP-AMP synthetase, IFN /: interferon /, IFNAR1: interferon alpha and beta receptor subunit 1, IFNAR2: interferon alpha and beta receptor subunit 2, JAK1: Janus kinase 1, IRF3: interferon regulatory factor 3, IRF9: interferon regulatory factor 9, ISGs: interferon-stimulating genes, ISG15: interferon-stimulating gene 15, MDA5: melanoma differentiation-associated gene 5, MAVS: mitochondrial antiviral signaling 5, POLA1: DNA polymerase 1, PSMB4: proteasome subunit beta 4, PSMB8: proteasome subunit beta 8, PSMA3: proteasome subunit beta 3, RNASEH2A: ribonuclease H2A, RNASEH2B: ribonuclease H2B, RNASEH2C: ribonuclease H2C, RIG-1: retinoic acid inducible gene-1, SAMHD1: SAM domain and HD domain containing deoxynucleoside triphosphate triphosphohydrolase 1, SKIV2L: superkiller viralicidic activity 2-like RNA helicase, STAT1: signal transducers and activators of transcription 1, STAT2: signal transducers and activators of transcription 2, TYK2: tyrosine kinase 2. |
Table 1.
The monogenic autoimmune diseases with corresponding pathogenesis and phenotype
Table 2.
Differences in clinical manifestations of IL-1 and IFN-mediated autoinflammatory diseases
IL-1: interleukin-1, IFN: interferon, CANDLE: Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature, SAVI: STING associated vasculopathy with onset in Infancy, AGS: Aicardi-Goutieres syndrome, CRP: C-reactive protein, CNS: central nervous system, MSK: musculoskeletal system, ENT: ear, nose, throat. This table is adapted from Kim et al. J Mol Med (Berl) 2016;94:1111-27 [13].
 XML Download
XML Download