

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Transthyretin (TTR) amyloidosis is a life-threatening systemic type of amyloidosis caused by an autosomal dominant hereditary mutation of TTR. TTR mutation leads to destabilization of its tetrameric structure and the aggregation of misfolded TTR monomers in numerous tissues including the peripheral nerves, heart, and eyes.1 The most-common TTR mutation is a substitution of methionine for valine at position 30 (Val30Met), with endemic foci reported in Portugal, Japan, and Sweden.234
TTR amyloidosis can have a heterogeneous presentation as polyneuropathy predominant (known as familial amyloid polyneuropathy), cardiac disease predominant (known as TTR cardiomyopathy), or mixed type. A recent observational study suggested that there is a correlation between the genotype (mutation type) and phenotype in TTR amyloidosis.56
Few cases of TTR amyloidosis have been described in South Korea.789 Because the Korean population has a relatively uniform genetic background and spatial proximity to other countries in East Asia, it was assumed that the epidemiological characteristics of TTR amyloidosis in Korea would be similar to those in Japan. Nevertheless, a recent molecular genetic analysis of seven families with TTR mutations suggested that the predominant mutation type differs between the South Korean and Japanese populations.210 Large epidemiological studies investigating TTR mutations in Korea have not been conducted due to the rarity and heterogeneity of TTR amyloidosis in this population.
In the present study we reviewed 18 patients from unrelated families who harbored TTR mutations, and analyzed their presentation and geographical distribution in South Korea.
METHODS
A retrospective cross-sectional study of all patients diagnosed with TTR amyloidosis was performed at two tertiary medical centers in Seoul, Korea. The collaborative investigation of medical records at these two medical centers in this study was approved by the representative ethics committee (IRB No. KUH1170161). Given the retrospective nature of the study and the use of anonymized patient data, the requirement to obtained informed consents was waived. Both centers received patients referred by other medical centers in Korea for diagnosing and managing hereditary TTR amyloidosis. The included patients had a diagnosis of systemic amyloidosis, clinical symptoms such as amyloid neuropathy or cardiomyopathy, and confirmation of a TTR mutation using genetic analysis recorded between April 1995 and November 2014.
Detailed patient and family-member histories were taken by one of the investigators. The geographical distribution of the affected family members was also investigated. The age at symptom onset was reported by the patient as the age at which they first experienced symptoms associated with TTR amyloidosis. Subjects with a symptom onset at younger than 50 years were classified as early onset, while those with symptom onset at 50 years of age or older were classified as late onset. The age at diagnosis was the age when a diagnosis of TTR amyloidosis was confirmed by genetic analysis. Symptoms including paresthesia, hypesthesia, and weakness with a length-dependent pattern were classified as peripheral neuropathy. Autonomic symptoms consisted of orthostatic dizziness, abnormalities in sweating, genitourinary dysfunction (e.g., impotence or urinary retention), and fecal incontinence.
Special attention was paid during the neurological examination to the distribution of sensory deficits and the severity of muscle weakness. Nerve conduction studies were performed to accurately diagnose polyneuropathy and to evaluate the disease severity. All subjects were evaluated by a neurologist using the modified polyneuropathy disability (mPND) score as follows: 0, normal walking; I, sensory disturbance in lower limbs but preserved walking ability; II, difficulties in walking but not needing a walking aid; IIIa, walking with one cane or crutch; IIIb, walking with two canes or crutches; and IV, using a wheelchair or confined to bed.11
Screening tests for autonomic dysfunction (heart-rate variability during deep breathing and the Valsalva maneuver, headup tilt testing, and quantitative sudomotor axon reflex testing) were performed to clarify the nature of vague symptoms related to autonomic neuropathy (e.g., fatigue and dizziness) and the existence of small-fiber neuropathy caused by amyloidosis. Symptoms of cardiac disease included exertional dyspnea, palpitation, or chest discomfort, and the results of screening tests including echocardiography and Holter monitoring. Autonomic dysfunction and amyloid deposition in the gastrointestinal (GI) tract directly causing severe nausea and diarrhea were noted as among the most-prominent characteristic symptoms. When reported, these symptoms were classified as GI symptoms and confirmed using an endoscopy examination to detect amyloidoma and exclude other causes of diarrhea.
These clinical evaluations were used to classify subjects into one of the following three phenotypes: 1) neurological type, with walking disability or other neurological symptoms of any severity, or GI symptoms and no cardiac abnormalities, 2) cardiac type, with abnormal electrocardiograms and only mild neurological abnormalities (mPND score 0 or I) or GI symptoms, and 3) mixed type, which covered all remaining symptomatic subjects who did not meet the criteria for either the cardiac or neurological phenotype. Other types of systemic amyloidosis were excluded by laboratory testing using protein immune electrophoresis of the serum and urine. Point mutation of TTR was identified by direct sequencing of all TTR coding exons and flanking intronic regions on the ABI Prism 3730 Genetic Analyzer using the Big Dye Terminator cycle Sequencing Ready Reaction Kit; Promega, Madison, WI, USA.
RESULTS
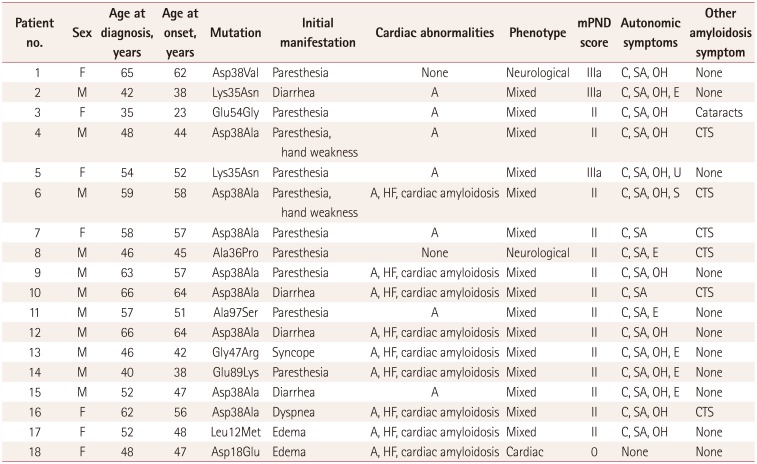
Eighteen proband cases of hereditary TTR amyloidosis (11 men and 7 women from unrelated families) were identified (Table 1). The mean age at disease onset was 49.6 years (range, 23–64 years) and the mean disease duration from symptom onset to diagnosis was 3.67 years (range, 1–12 years). Nine patients (nos. 1, 5–7, 9–12, and 16) were in the late-onset group, and six of them harbored the Asp38Ala mutation. The other three late-onset patients (nos. 1, 5, and 11) were genotyped as Lys35Asn, Asp38Val, and Ala97Ser, respectively.91213
Seventeen patients presented symptoms of peripheral neuropathy and had electrophysiological findings consistent with axonal sensorimotor polyneuropathy. Six patients (nos. 4, 6–8, 10, and 16) had comorbid severe carpal tunnel syndrome (CTS) with compatible findings in the nerve conduction study, which indicated that the symptoms of neuropathic pain had started in the bilateral upper limbs. Five of the six patients with CTS harbored the Asp38Ala mutation.14
The presence of cardiac disease related to a conduction defect or decreased ejection fraction was confirmed in 16 patients. All of these patients had limitations in performing the activities of daily living due to generalized edema, exertional dyspnea, chronic fatigue, or palpitations. Autonomic nervous abnormalities were also common clinical features, with 17 of the 18 patients presenting with orthostatic hypotension, urinary retention, impotence, or abnormal sweating.
Their GI symptoms primarily included severe diarrhea with weight loss, which were reported as the first clinical presentation in four patients (nos. 2, 10, 12, and 15). One of these patients (no. 15) complained of diarrhea as the main symptom of amyloidosis, and subsequent pathological findings confirmed amyloid deposition in the GI tract. Only one patient (no. 3) was treated surgically due to cataracts related to amyloid deposition. Sixteen of the 18 patients underwent tissue biopsies for pathological evaluations, which confirmed amyloid deposition in the nerves, fat, GI tract, or heart.
A genetic analysis detected Asp38Ala—the most-common mutation pattern—in eight patients (nos. 4, 6, 7, 9, 10, 12, 15, and 16). The mean age at onset in these patients was 55.9 years, with early onset in only two of these patients (nos. 4 and 15, at 44 and 47.2 years of age, respectively). Lsy35Asn was detected in two patients (nos. 2 and 5) from unrelated families. The remaining eight patients harbored different mutation types. Eight patients with Asp38Ala had similar clinical symptoms, such as severe axonal polyneuropathy, cardiac disease, and autonomic neuropathy, and three patients (nos. 10, 12, and 15) with Asp38Ala complained of severe diarrhea with weight loss. The only patient with significant ocular symptoms due to amyloid deposition (no. 3) was genotyped as Glu54Gly.7 Two patients with Lys35Asn also shared similar symptoms including sensorimotor polyneuropathy and autonomic dysfunction.9
Fifteen of the 18 patients were classified as the mixed phenotype and 2 (nos. 1 and 8) were the neurological phenotype. The only patient (no. 18) with the Asp18Glu mutation was classified as the cardiac phenotype, mainly presenting with exertional dyspnea and edema and without neurological abnormality.2
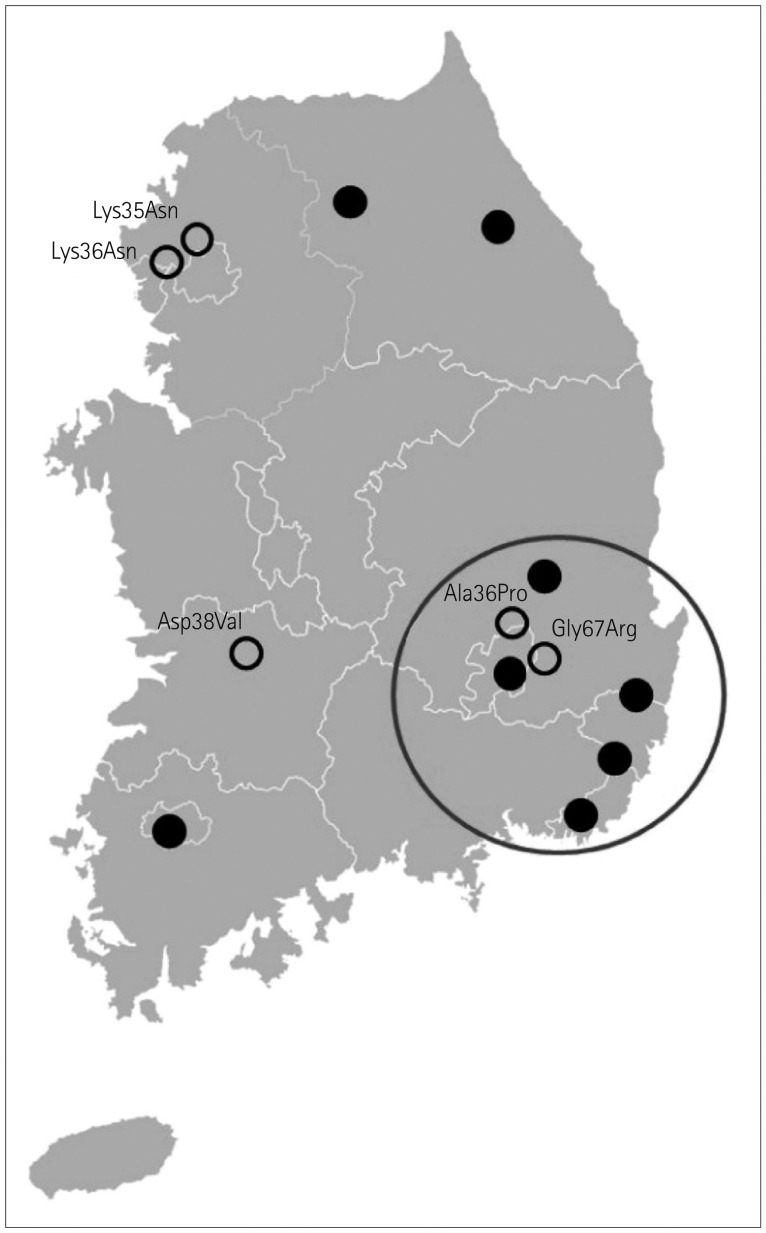
Finally, with regard to geographical distribution, 13 patients reported their family hometowns (Fig. 1). Five of the eight patients harboring an Asp38Ala mutation were from the Gyeongsang province, which is in southeast Korea. Two patients harboring the Lys35Asn mutation were from Paju city;9 the remaining eight exhibited a widespread geographical distribution.
DISCUSSION
TTR amyloidosis due to TTR mutation was historically considered to be a rare endemic disease, but recent epidemiological studies using improved diagnostic methods have suggested that it is more widely distributed than previously believed.2151617 The present South Korean patients with TTR amyloidosis exhibited heterogeneous TTR genotypes and clinical phenotypes. TTR amyloidosis was classified into three main clinical phenotypes (neurological, cardiac, and mixed), and our patients mainly presented with the mixed type (15 of 18 patients). Peripheral neuropathy is the most-common clinical presentation of TTR amyloidosis, and has been strongly associated with the most-common TTR mutation type of Val30Met.18 In the present epidemiological study, sensorimotor polyneuropathy was the predominant symptom of TTR amyloidosis in patients across different mutation types: Asp38Ala, Ala97Ser, Lys35Asn, and Asp38Val.9121314
We found cardiomyopathy to also be a common clinical feature in TTR mutations, with this being absent in only two patients (with mutation types Asp38Val and Ala36Pro).1419 Previous studies have similarly indicated that cardiac presentation is not a characteristic symptom of Ala36Pro, because this mutation has largely been associated with polyneuropathy, vitreous opacity, and leptomeningeal amyloidosis.192021 However, a recent study involving Chinese patients with TTR amyloidosis identified rapidly progressing cardiomyopathy as a feature of disease associated with the Ala56Pro mutation.22 Val122Ile and Thr60Ala have been the mutation types most commonly associated with TTR cardiomyopathy, especially in African-American populations;2324 however, these mutations were not found in the present study.
Cardiomyopathy and autonomic neuropathy are the largest contributors to sudden death in patients with TTR amyloidosis. Sixteen of our patients reported symptoms of autonomic dysfunction. Autonomic neuropathy tends to develop early in patients with TTR amyloidosis and can present as erectile dysfunction (in men), urinary retention, dysregulation of orthostatic blood pressure, or abnormal sweating. An autopsy study found pathogenic amyloid deposits and nerve injury in several patients harboring the Val30Met mutation, with heavy aggregation detected in the spinal ganglia and nerve roots.25
The most-common TTR mutation in our South Korean patients was Asp38Ala. Previous clinical characterizations of the Asp38Ala mutation have described prominent heart disease, GI tract involvement, polyneuropathy, and autonomic neuropathy in Japanese studies.1426 Eight patients harboring Asp38Ala in our study presented with a clinical phenotype similar to that reported previously. Notably, five of these eight patients had confirmed severe CTS and presented with initial neuropathy symptoms in the upper extremities (Table 2). It is noteworthy that we did not detect the Val30Met mutation in our South Korean patients given that this mutation was previously reported as the most-common TTR mutation worldwide, including in Japan. Nevertheless, the most-common mutation in our study (Asp38Ala) was found in five Korean patients from the Gyeongsang province (Fig. 1). Yamashita et al.27 reported that the prevalence of non-Val30Metmutation TTR amyloidosis in Japanese patients was higher than expected, and the diversity of their clinical presentations was similar to that in the present study. These outcomes suggest that the distribution of TTR amyloidosis in South Korea may be due to de novo mutations and/or related to the Japanese population.
This study was subject to some limitations. First, patients with wild-type TTR amyloidosis were not enrolled, and so the evaluation of phenotypic characteristics was limited to TTR amyloidosis caused by TTR mutations. Second, not all of the included patients had been diagnosed during the early stage of disease, and so the reported clinical presentations might not have completely reflected the actual genotype-phenotype relationships. Third, we were able to enroll only a small number of patients; a larger sample would provide morereliable information about the characteristic geographical distributions of different mutations in South Korea.
Future studies should investigate the early-stage clinical manifestations of TTR mutation types in South Korea. In particular, the healthy offspring and relatives of patients with the Asp38Ala mutation should be investigated with the aim of understanding their natural course by using long-term observations. Additionally, a better characterization of geographical distribution may aid the development of early diagnostic tests for identifying asymptomatic carriers of TTR mutations.
 XML Download
XML Download