

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Paroxysmal dyskinesia is a rare and heterogeneous group of movement disorders characterized by recurrent episodes of involuntary dystonia, chorea, athetosis, or their combination. It is usually categorized into three subgroups based on the triggers: paroxysmal kinesigenic dyskinesia (PKD), paroxysmal nonkinesigenic dyskinesia (PNKD), and paroxysmal exercise-induced dyskinesia (PED).12 Hypnogenic paroxysmal dyskinesia, formerly considered to be the fourth type of paroxysmal dyskinesia, is now known to be a form of nocturnal frontal lobe epilepsy.3 PKD, the most common type of paroxysmal movement disorder, shows brief recurrent attacks precipitated when initiating voluntary movements. The duration of an attack is usually shorter than for other types, commonly lasting for less than 1 min. The age at onset is usually between 7 and 15 years, and patients show fair responses to anticonvulsants such as carbamazepine or oxcarbazepine.45 Attacks in patients with PNKD usually have a longer duration (up to several hours) and are triggered by alcohol, coffee, or emotional stress.6 Symptoms usually begin before the age of 20 years and treatment responses are worse than in PKD. Symptoms of PED are triggered by sustained exercise, with a mean age at onset of 5 years, and anticonvulsant therapy does not seem to be very useful.4 All three types of dyskinesias are often associated with other neurologic disorders such as infantile seizures, other types of epilepsy, migraine, writer's cramp, ataxia, or tremor.46789101112
Since the myofibrillogenesis regulator 1 gene (MR-1, OMIM 609023) was first identified as the cause of PNKD in 2004,13 protein-rich transmembrane protein 2 gene (PRRT2, OMIM 614386) for PKD and solute carrier family 2, member 1 gene (SLC2A1, OMIM 138140) for PED have been found.1415 Although recent studies have found that the phenotype and genotype overlap as well as various manifestations within the subtypes and phenotype heterogeneity over the paroxysmal dyskinesias,161718 there have been few large integrated studies, especially those involving the pediatric population. We therefore aimed to determine the clinical and genetic features of pediatric patients with paroxysmal dyskinesias and identify the clinical and genetic heterogeneities.
METHODS
Study approval and registration of patients
The study was approved by the Institutional Review Board of Seoul National University Hospital (IRB No. 1101-110-353 for DNA preparation and IRB No. 1708-148-879 for genetic tests and the retrospective review of medical records). All of the patients or their legal representatives provided written informed consent to participate in the present study.
Sequencing for PRRT2, SLC2A1, and MR-1
Genomic DNA was extracted from peripheral blood leukocytes using a QIAamp DNA Blood Midi Kit according to the manufacturer's instructions (Qiagen, Valencia, CA, USA). Polymerase chain reaction (PCR) amplification was performed with each specific primer pair designed by the authors. The PCR conditions used were 95℃ for 3 min, followed by 35 cycles of 95℃ for 30 s, 57℃ for 30 s, and 72℃ for 40 s, with a final extension at 72℃ for 5 min. Sanger sequencing reactions were run on an ABI 3730XL DNA Analyzer (Applied Biosystems Inc., Carlsbad, CA, USA) to identify PRRT2, SLC2A1, and MR-1. After PRRT2 sequencing was performed for all patients, SLC2A1 sequencing was performed for the remaining PRRT2-negative patients. Thereafter, Sanger sequencing of MR-1 was performed for mutation-negative patients with PNKD. The pathogenicity of variants was evaluated according to the standards and guidelines of the American College of Medical Genetics and Genomics (ACMG).20
Data collection and analysis
We reviewed the medical records of all patients to obtain information on age, sex, symptoms, age at onset, family history, combined medical conditions, medication, and responsiveness to treatment. We classified patients into three groups according to clinical criteria4619 and described the clinical features of all of the patients. Treatment responses were classified into the following four categories: symptom-free, markedly improved (75–99% reduction of symptoms), minimally improved (25–75% reduction of symptoms), and no change (0–24%) or aggravated. We compared the clinical features between patients with PKD and PNKD, and furthermore analyzed those of PKD patients with and without PRRT2 mutations.
Statistical analyses were conducted using the IBM SPSS statistics version 22.0 software suite (IBM Corp., Armonk, NY, USA). Student's t test was applied to continuous variables and the Pearson chi-square test applied to categorical variables. The range of probability values for statistical significance was set at p<0.05.
RESULTS
Clinical characteristics
Among the 55 cases in this study, 40 (72.7%) were categorized as PKD, 14 (25.5%) as PNKD, and only 1 (1.8%) as PED. Seventeen cases (30.9%) were familial: 13 with paroxysmal dyskinesia and 4 with benign familial infantile seizures (BFIS). Fifteen cases from 13 families were categorized as PKD, 1 as PNKD, and 1 as PED. Males comprised 38 (69.1%) of the patients. The follow-up duration was 3.20±3.27 years (mean±SD, range=0.2–14 years), and the age at symptom onset was 8.80±4.53 years (range=0.4–15 years). Brain MRI was performed in 49 patients (89.1%), with the findings being unremarkable except for 1 case of venous angioma and 1 of dysgenesis of the corpus callosum.
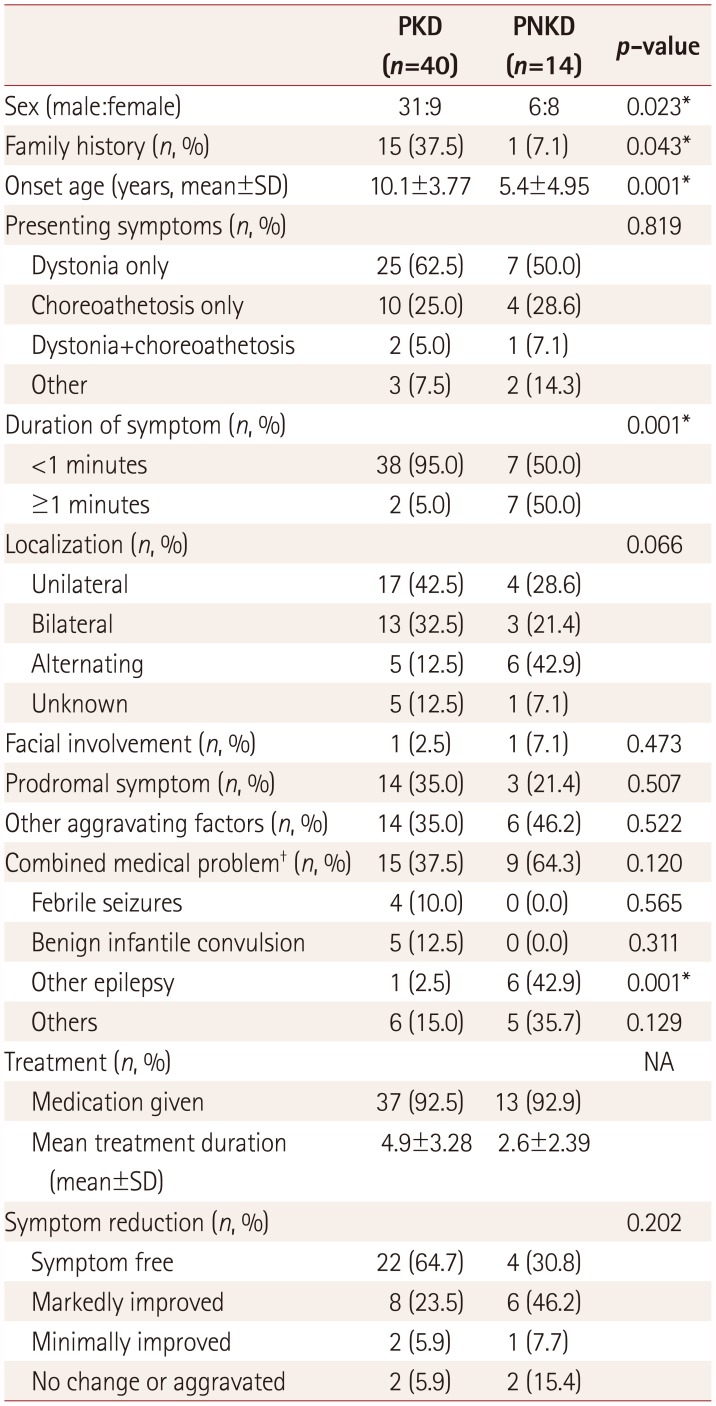
The clinical characteristics of PKD and PNKD are compared in Table 1. The most common symptom in both groups was dystonia. Patients with PKD were more likely to have a family history (p=0.043) and shorter symptom attacks (p=0.001). Patients with PNKD presented their first symptom at an earlier age (p=0.001). Epilepsy other than BFIS were more commonly found in patients with PNKD (p=0.001), while BFIS were observed only in patients with PKD. Comorbidities such as mild intellectual disability, tic disorder, and attention deficit hyperactivity disorder (ADHD) were observed in 6 patients with PKD (15%) and 5 with PNKD (35.7%). Fourteen patients with PKD (35.0%) had other additional triggering factors such as tension, emotional stress, cold exposure, and infections. Antiepileptic drugs (including oxcarbazepine) were administered to 37 and 13 patients in the PKD and PNKD groups, respectively, and a symptom-free state was achieved in 22 (64.7%) and 4 (30.8%) patients, respectively.
Genetic analysis of PRRT2, SLC2A1, and MR-1
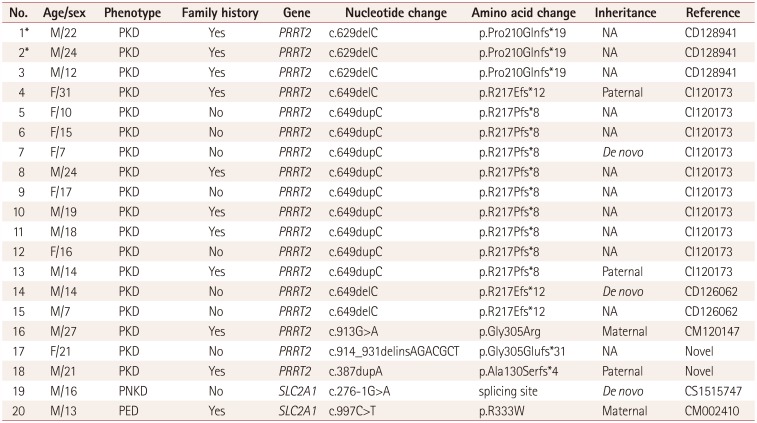
Pathogenic variants in PRRT2 and SLC2A1 were identified in 18 and 2 patients, respectively, while no MR-1 mutations were found. The phenotypes and genotypes of all mutation-positive patients are described in Table 2. All PRRT2-positive patients presented as PKD alone, whereas two patients with SLC2A1 mutations had PNKD and PED. All of the PRRT2 mutations were frameshift mutations, with the exception of a single missense mutation. The c.649dupC variant in PRRT2 was the most common (9/17, 52.9%). Additionally, two novel mutations in PRRT2 were identified: c.387dupA and c.914_931delinsAGACGCT, both of which were pathogenic based on the ACMG standards and guidelines. The 2 patients with SLC2A1 mutations comprised one with a splice-site mutation and one with a missense mutation: c.276-1G>A and c.997C>T.
Comparison between PKD patients with and without PRRT2 mutations
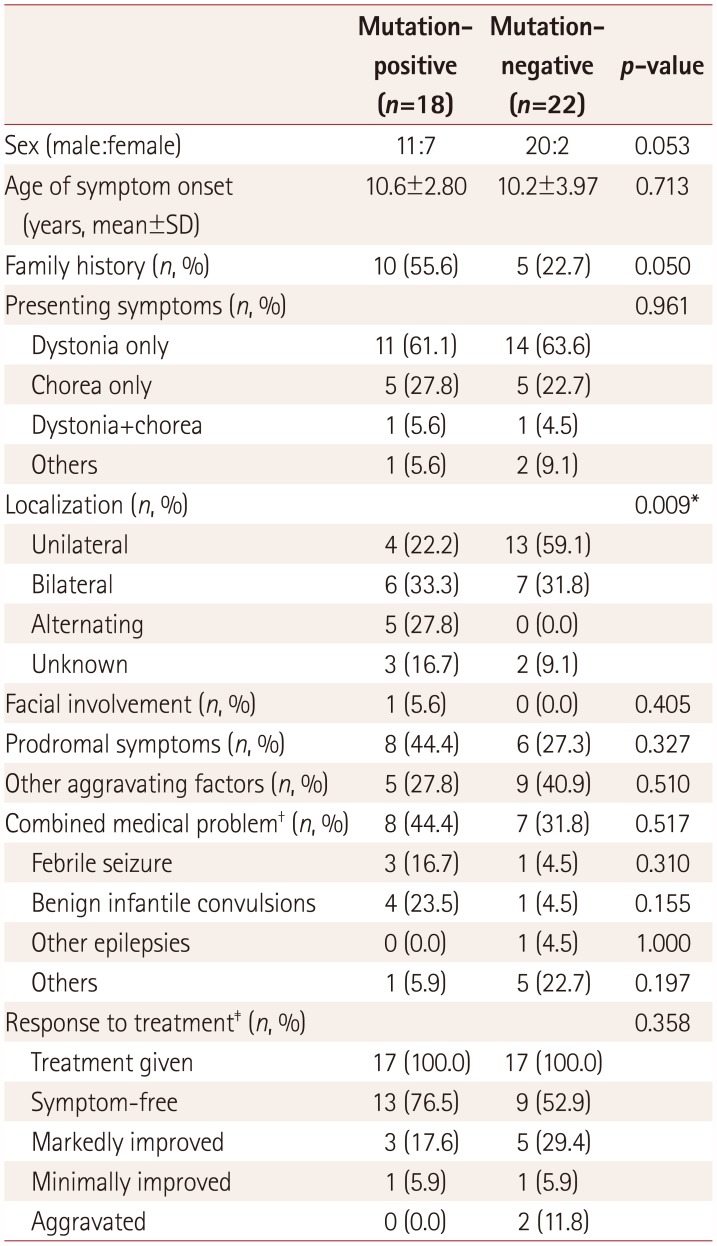
We compared the clinical features between PKD patients with and without PRRT2 mutations (Table 3). Familial cases were more common in PKD patients with PRRT2 mutations (p=0.050). Dyskinesia of both sides of limbs manifested in 61.1% of the PRRT2-positive patients, with the remainder showing unilateral dyskinesia (p=0.009). No significant differences were observed in the main presenting symptom or combined medical problems, although PRRT2-positive PKD patients were more likely to have a history of BFIS (p=0.155), and PRRT2-negative PKD patients were more likely to have other medical problems such as tic disorder, ADHD, or intellectual disability (p=0.197). Both groups showed good treatment responses: 94.1% and 82.3% in PRRT2-positive and PRRT2-negative PKD patients, respectively.
DISCUSSION
Paroxysmal dyskinesia can be classified into three groups based on the triggers, and this classification facilitates the diagnosis. However, clinical and genetic heterogeneity (even within the same phenotype) as well as phenotype–genotype overlap among the three groups reportedly make it difficult to establish an etiologic diagnosis in some cases. Additionally, the pathophysiologies of PKD, PNKD, and PED associated with the roles of PRRT2, SLC2A1, and MR-1 have not been elucidated completely, even though recent studies on synaptopathies, transportopathies, and channelopathies have been reported.2122
The clinical and genetic characteristics were investigated in this large cohort of pediatric patients with paroxysmal dyskinesia. As expected, PKD was the most common type and showed a male predominance with a mean age at onset of 10.1 years. Dystonia was the most common symptom in these patients, with short duration of less than 1 min, and 64.7% achieved a symptom-free state with oxcarbazepine or carbamazepine alone. Five patients with PKD (12.5%) had a history of BFIS, four of whom carried PRRT2 mutations. Six PRRT2 mutations were identified in 9 of the 13 tested families (69.2%) and in 8 patients of the 25 tested sporadic cases (32.0%) of PKD. The most common mutation was c.649dupC, which accounted for 50% of PRRT2-positive PKD patients. This mutation was not carried in sporadic cases in another Korean cohort, whereas we identified it in 62.5% of patients with sporadic cases of PRRT2-positive PKD.23 This represents a hotspot mutation in patients with sporadic and familial cases of PKD worldwide.1624 Consistent with previous studies, 35% of all of the patients with PKD (about 22% of PRRT2-positive PKD patients) had other additional triggering factors besides initiating voluntary movements, including tension, emotional stress, cold exposure, and infections. The treatment response did not differ significantly between the PRRT2-positive and PRRT2-negative groups, which is consistent with previous studies and might be due to some pathomechanisms being shared between different genetic defects.2325
The second most common type in this study was PNKD, and all but one of the cases were sporadic. Only one of the patients with PNKD had variants in the three genes: a pathogenic variant in SLC2A1. Most studies of PNKD reported to date have involved familial cases, and furthermore there have been no sporadic cases with MR-1 mutations reported.262728 A recent study indicated that the mean age at symptom onset among patients with MR-1-negative familial PNKD was over 12 years.26 Our patients with PNKD showed their first symptom at a mean age of 5.4 years. Moreover, their attacks tended to be shorter, with a duration of only a few seconds in about half of our patients, compared with this exceeding 10 min in previous reports.262728 Although our patients were clinically classified into the PNKD type based on the triggers, they might represent another group within pediatric paroxysmal dyskinesia rather than simply MR-1-negative PNKD. As expected, other comorbidities such as epilepsy, tic disorder, ADHD, or cognitive disability were commonly reported in our PNKD patients.26 Together these findings indicate that neurologic comorbidities such as neurodevelopmental delay or epilepsy other than BFIS are observed in mutation-negative patients with PKD or PNKD, and suggest the presence of other genetic mutations such as a channelopathy in SCN8A or KCNMA1. Further investigations that include genetic testing for these two genes might be needed in the remaining mutation-negative patients.
Phenotype–genotype overlap was identified in the present study, although there were no PRRT2-positive patients presenting with PNKD or PED. One patient with a splicesite mutation (c.276-1G>A in SLC2A1) showed PNKD, and another patient carrying the c.997C>T variant had PED. Both of these patients exhibited developmental delay and epilepsy. It is known that SLC2A1 mutations are usually found in PED patients, but the PNKD phenotype has also been reported.16 Our SLC2A1-positive PNKD patient experienced dystonic attacks that typically lasted 10–15 min, occurred frequently during the day, and were triggered by emotional stress, sustained exercise, and fatigue. He was diagnosed with glucose transporter 1 (GLUT1) deficiency after a low glucose level was measured in the cerebrospinal fluid (37 mg/dL; ratio of cerebrospinal fluid to serum glucose of 0.3), and showed marked improvement of symptoms after consuming a ketogenic diet. The other patient with the c.997C>T variant in SLC2A1 was a 13-year-old boy who suffered from intermittent dystonia in his lower extremities triggered by intense exercise or rapidly walking 700–800 meters. We diagnosed GLUT1 deficiency and started him on diet therapy. Although SLC2A1 mutations accounted for a small portion in our cohort, the elucidation of genetic causes is very important for initiating early treatments.
Most previous investigators have reported the clinical features of PKD patients as well as the genetic analysis of PRRT2, with mutations reportedly appearing in from 18.5% to 65% of cases.232425 One recent study found PRRT2, SLC2A1, and MR-1 mutations in 35%, 10%, and 2% of 145 patients with paroxysmal dyskinesia, respectively, indicating clinical and genetic heterogeneity, as well as phenotype–genotype overlap.16 There have been no genetic or clinical studies involving a large cohort of patients with pediatric paroxysmal dyskinesia. We screened for the top-3 causative genes in 55 patients from 53 families with pediatric paroxysmal dyskinesia, although sequencing for MR-1 was only performed in patients with PNKD. Only one of the PRRT2-negative and SLC2A1-negative patients with paroxysmal dyskinesia had an autosomal dominant family history in our present cohort that had PKD with epilepsy, and therefore a lower possibility of any MR-1 mutations. PRRT2, SLC2A1, and MR-1 mutations were identified in 18 (32.7%), 2 (3.6%), and 0 of the present cohort with pediatric paroxysmal dyskinesia patients, respectively.
In conclusion, the present study has expanded the clinical and genetic spectrum of pediatric paroxysmal dyskinesia. Additionally, both the heterogeneity and phenotype–genotype overlap of paroxysmal dyskinesia have been summarized. In some cases it is difficult to make a diagnosis based on clinical features alone. This means that broader genetic testing including for at least three genes should be performed in patients with pediatric paroxysmal dyskinesia to aid the search for treatable cases.
 XML Download
XML Download