

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Prune belly syndrome (PBS) is a rare and usually a fatal congenital condition which presents with a partial or complete triad of deficient or absence of abdominal musculature, cryptorchidism and urinary tract anomalies [123]. Other congenital anomalies such as pulmonary hypoplasia, potters facies, clubfoot, imperforate anus are also found to be associated with PBS [4]. The incidence of this genetic disorder is seen among 1 in 29,000–50,000 live births usually affecting males [15]. In 1839, the absence of abdominal musculature was first described by Frolichs which was later associated with urinary tract anomaly by Parker in 1875 [126]. Genetically normal karyotyping is observed in most of the case of PBS [5]. However, Younous et al. [3] and Metwalley et al. [7] had reported PBS to be associated with Down's syndrome and other malformations like Vacteryl. Also, the chromosome 17q 12 deletions encompassing hepatocyte nuclear factor 1-β (HNF 1-β) can be determinate in cases of PBS [8].
Case Report
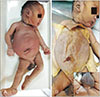
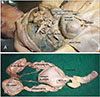
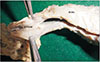
Full-term fetus embalmed and fixed in formalin was selected for exploration, as the fetus appeared to have some gross anomaly externally. A detailed external examination of the fetus was done before exploring the interior. The fetus exhibited potters facies and unusually large and distended abdomen. The external genitalia showed the scrotum devoid of the testis. The penis appeared larger than usual (mega penis) (Fig. 1A). The anal opening was absent in the fetus (imperforate anus) (Fig. 1C). The lower limb presented bilateral club feet. With the curiosity to know the details, it was decided to explore the interior of the fetus; the abdominal cavity was exposed by reflecting the abdominal layers. On reflection of the skin of the abdomen, the abdominal muscles were present (Fig. 1B). However, the extent of hypoplasia of these muscles could not be examined visually. Further, the anterior abdominal wall was reflected distally at the level of costal margins. The peritoneal cavity was opened, and each abdominal organ was examined to rule out the anamoly. We found abnormality in the urinary tract, unusually large bladder (mega cystitis), bilateral hydroureter with dilated and thin-walled ureters and bilateral hydronephrosis (Fig. 2). To rule out the anomaly in the interior, the bladder was opened, and the ureteric orifices were present at the usual site, and the urethral orifice also appeared normal and patent. The mega penis was opened by slitting sagittal to study the anatomy of the urethra. The penile part of urethra showed canalization. While on further exploration, it was observed that the prostatic part of the urethra was not canalized (Fig. 3).
On further exploration, small and underdeveloped testis was observed close to the lateral abdominal wall lateral to the ureter, which failed to descend to the scrotum (Fig. 2A).
The proximal and middle part of the gastrointestinal tract (stomach and small intestines) appeared normal. However, the distal part the tract appeared underdeveloped. On further tracing, the sigmoid colon was distended and filled with fecal matter/meconium, and the rectum was observed to be narrow which did not continue distally. Instead it formed an atretic cord and appeared to be connected to the median lobe of prostate as fibrotic connective tissue (Fig. 4A, B). On exploring the thoracic cavity, the thoracic organs were located in anatomical positions, but the lungs appeared smaller than usual suggesting pulmonary hypoplasia.
Discussion
PBS is also named as Eagle-Barrett syndrome. It is a morbid congenital disease with higher incidences of mortality. The prognosis of PBS is poor with cases of stillbirths or deaths within first few weeks after birth [9]. The etiology and pathogenesis of PBS are not understood. Various theories have been proposed for the cause of PBS. One of the theory explains the prenatal obstruction in urinary tract as the cause for PBS leading to megaureter, hydronephrosis, abdominal distension thereby causing abdominal muscle weakness and cryptorchidism [1810]. Failure in mesoderm differentiation derived from first lumbar myotome is proposed to be the secondary theory for PBS which leads to defective musculature in both abdominal and urinary musculature [14810]. Lastly, dysgenesis of yolk sac and allantois could also be a cause for PBS [10]. However, Fette [1] suggests that each theory explains some components of PBS and fails to explain the syndrome completely. For instance, cases have been reported where the posterior urethral valve in infants shows similar primary clinical findings and features as that of PBS, but abdominal muscle deficiency would not be seen in such cases [16]. Histology of bladder in PBS and posterior valve obstruction showed thickness in detrussor muscle in latter and more connective tissue ratio in the former [6]. Urinary tract obstruction leads to secondary manifestations of oligohydramnios, pulmonary hypoplasia, potters facies and clubfoot in PBS [7].
Although the genetic basis for PBS is not understood, few studies have been reported on the same. Ramasamy et al. [11], 2005 strongly believes that sex influenced autosomal recessive mode of inheritance as the likely cause for PBS. strongly believes that sex influenced autosomal recessive mode of inheritance as the likely cause for PBS. A large interstitial deletion of long arm of chromosome 6 was diagnosed in a male fetus with early urethral obstruction and PBS [12]. PBS was also reported in a 4-month-old Egyptian boy with Down's syndrome [7]. A de novo case 1.3 megabase interstitial 17q12 microdeletion including HNF1-β gene was reported [8]. The normal mesodermal and endodermal development including kidney, prostate, mesonephric duct derivatives, pancreas, gut, and liver is regulated by expression of a HNF1-β transcription factor. However, a cohort study of 32 cases with PBS, only a single case showed mutation of HNF1-β V61G (valine at amino acid position 61), hence the authors claim that further additional candidate gene must be identified to understand the genetic basis of PBS [13]. A review literature on molecular regulation of kidney development reports that inactivation of signalling protein shh in collecting duct epithelium ends up in development of renal hypoplasia, hydronephrosis and hydroureter [14].
In the present case, primary clinical findings of PBS are observed. The abdomen appeared large; however, the abdominal muscle was present. But the extent of hypoplasia or deficiency of musculature could not be appreciated visually. Though deficient abdominal musculatures one of finding in PBS, it was not understood in our case. To rule out the provisional diagnosis, i.e., posterior urethral valve obstruction, the urethral orifice was observed, and it appeared patent. Moreover, the present case also showed features such as imperforate anus, club feet, and gastrointestinal malformations. Further, the present case correlates with a retrospective study of Bellah eand his co-workers that reported the existence of pseudoprunes with normal abdominal wall examination, PBS uropathy and complete or partial cryptorchidism [15]. that reported the existence of pseudoprunes with normal abdominal wall examination, PBS uropathy and complete or partial cryptorchidism.
Though the complete triad of PBS was not seen in our case, with the presence of abdominal musculature, all the secondary characters were associated. So we consider our case to be pseudoprune with partial manifestations of the triad. Many cases of PBS are still born or viable only for few weeks. However, prenatal detection of PBS is crucial, through sonographic examination and may help to reduce the complications of the disease by proper planning of the interventions to be carried out antenatally and thereby prevent the rate of morbidity.

 XML Download
XML Download