

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Human mitochondrial DNA (mtDNA) is a circular molecule of 16,569 bp that contains 37 genes coding for 13 polypeptides of the mitochondrial electron respiratory chain, two ribosomal RNAs, and 22 transfer RNAs (tRNA) required for polypeptide synthesis [12]. In comparison to the nuclear genome, mtDNA has a modified genetic code, paucity of introns, and lack of histone protection [34]. Moreover, the turnover of mtDNA is high, as degradation and replication is a continuous process in the mitochondria during one cell cycle, and mtDNA polymerase does not have proofreading capabilities. Therefore, the accumulation of somatic mutations is greater in mtDNA compared to that in nuclear DNA [1]. The mtDNA molecule has a noncoding region, which includes a unique displacement loop (D-loop) responsible for replication and transcription control. This control region includes a mtDNA production regulation fraction and a hypervariable (HV) region known as a gene mutation “hot spot,” resulting in mtDNA sequence and length heteroplasmies [5]. The sequence heteroplasmies consist of base substitutions and small deletions and insertions. The length heteroplasmies are usually detected by varying numbers of a particular repeated nucleotide, usually poly-C, known as the mtDNA minisatellite instability (mtMSI) [6]. In conjunction with the control region, the tRNA
leucine 1 (tRNAleu1) and cytochrome b (CYTB) genes in the coding region represent approximately 13% of the total mtDNA sequence, and pathological heteroplasmic mtDNA mutations were observed in these regions of various cancer tissues [678].
The mitochondrion is an important micro-organelle that produces energy for cell development, differentiation, and growth. It also controls cell growth by inducing apoptosis. The mtDNA is comprised of 0.1% to 1.0% of the total DNA in most mammalian cells, and 2 to 10 copies come packaged in each nucleated cell per mitochondrion up to 1,000 mitochondria. The copy number per cell is controlled within a constant range to fulfill the energy requirement of the cell and to maintain normal physiological functions [910]. Variations in the mitochondria copy number have been reported to reflect the net results of gene-environmental interactions, and the copy number has been implicated as a potential biomarker for various cancers [1112].
Since mtDNA is haploid and lacks recombination, specific mutations in the mtDNA genome leading to human diseases arise in particular genetic backgrounds referred to as haplogroups [13]. Human populations usually carry several mtDNA haplogroups defined by unique sets of mtDNA polymorphisms, reflecting mutations accumulated by a discrete maternal lineage [1415], but the sets and their frequencies differ between populations. Thus, haplogroup association studies have been used to assess the role of mtDNA variants in various diseases and cancers [16].
Acute myeloid leukemia (AML) results from the accumulation of abnormal blasts in the bone marrow. It is likely that many different mutations, epigenetic aberrations, or abnormalities in microRNA expression can produce the same morphological disease; however, these differences may be responsible for the variable responses to therapy, which is a principal feature of AML [17]. In addition to these nuclear genetic changes, non-chromosomal mitochondrial mutations may play a role in the progression and chemosensitivity of leukemias [181920]. In recent years, mtDNA variants within the D-loop or the entire mitochondrial genome in patients with AML and their prognostic impacts have been reported [2122]. However, few studies have focused on the mtMSI, copy number, or haplogroups in association with AML.
In this study, we analyzed the control region and two coding regions in bone marrow cells of patients with AML to demonstrate the sequence and length heteroplasmies of mtDNA, alterations in mtDNA copy number, haplogroup distribution, and their impacts on patient outcomes.
MATERIALS AND METHODS
Patients and samples
Seventy-four patients with AML and 70 control subjects were enrolled in this study after obtaining Institutional Review Board approval and informed consent. The control subjects were selected from normal adults visiting the health examination center. The total DNA of bone marrow samples at the diagnostic stage from patients with AML was extracted with the AccuPrep Genomic DNA Extraction Kit (Bioneer, Daejeon, Korea). The BM specimens from the patients with AML were frozen in liquid nitrogen immediately after acquisition for further evaluation. The total DNA from the peripheral blood of control subjects was processed using the same protocol. The DNA from control subjects was used for detecting mtDNA sequence variation, measuring mtDNA copy number, and classifying haplogroups.
Direct sequencing of the mtDNA control and coding region
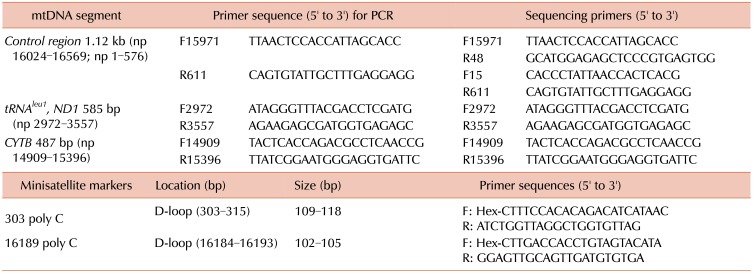
Using samples from AML patients and control subjects, we directly sequenced the control regions (HV1 and HV2), the tRNAleu1gene including a small portion of NADH dehydrogenase (ND1), and the cytochrome b (CYTB) gene of mtDNA, using a set of primer pairs according to our protocols (Table 1) [6]. The sequencing analysis was performed using the ABI Prism 3130XL Genetic Analyzer with the BigDye Terminator v3.1 Ready Reaction Kit (Applied Biosystems, Foster City, CA). To exclude potential artifacts, PCR amplifications from the original cell lysates were replicated once or twice and sequenced. The mtDNA sequences obtained were analyzed using the MitoAnalyzer (http://www.cstl.nist.gov/biotech/strbase/mitoanalyzer.html) with the Revised Cambridge Reference Sequence (http://mitomap.org/MITOMAP) and the Blast2 program (http://www.ncbi.nlm.nih.gov/blast/bl2seq/wblast2.cgi) to determine mtDNA aberrations.
Detection of mtDNA length heteroplasmy using minisatellite markers
Our previous protocols were used in order to examine the length of heteroplasmies using two mtDNA minisatellite markers 16189 poly-C (16184CCCCCTCCCC16193, 5CT4C) and 303 poly-C (303CCCCCCCTCCCCC315, 7CT5C) in the mtDNA HV1 and HV2 regions, respectively. The analysis was based on separation via capillary electrophoresis using an ABI Prism 3130XL Genetic Analyzer (Applied Biosystems) and Gene Scan Analysis Software (version 3.1, Applied Biosystems). TA cloning for the confirmation of mtDNA minisatellite alterations was carried out according to a previously published protocol [623]. We compared this mtMSI data of the patients with AML with our previously published data from healthy Korean donors [6].
Statistical analysis
The significance of observed differences in proportions was tested by Fisher's exact test. To analyze the AML risk, the odds ratio (OR) with 95% confidence interval (CI) was tested. The Mann-Whitney U test was used to determine significance between differences of the medians. Overall survival (OS) was calculated as the time from diagnosis until death from any cause, with the observation censored at the date of the last follow-up for patients last known to be alive. Event-free survival (EFS) was defined as the time from complete remission (CR) or bone marrow transplantation/peripheral blood stem cell transplantation (BMT/PBSCT) until relapse or death from any cause. In EFS analyses, only patients who achieved CR or who underwent BMT/PBSCT were included. Log-rank (Mantel-Cox) test was used to estimate OS and EFS. Cox regression analyses of specific parameters were performed to calculate univariate and multivariate P-values and hazard ratios (HR), considering patient characteristics such as age, white blood cell count, blast percentage, prognostic group based on cytogenetic study, and secondary AML. All statistical computations were performed using PASW 18.0 (SPSS, Chicago, IL, USA). P-value <0.05 was considered to indicate statistically significant differences.
RESULTS
Patient characteristics
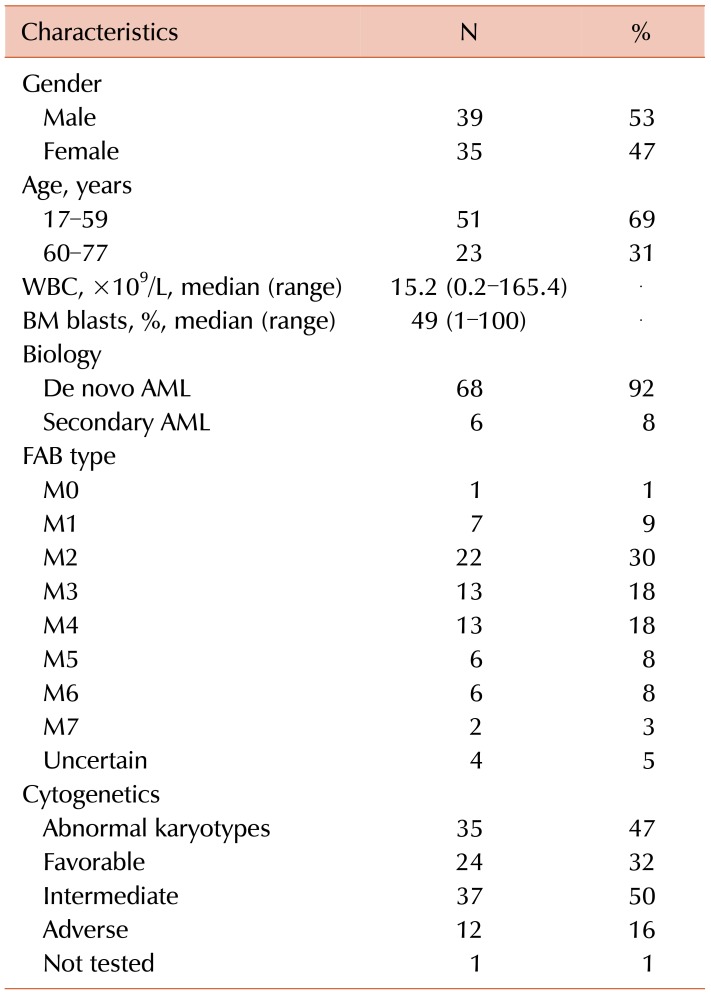
The patient cohort included diagnostic bone marrow samples from 74 adult patients with AML. The ages varied from 17 to 77 years, and the average was 48 years. The male and female ratio was 1.1:1. Six patients had secondary AML, and 68 patients had de novo mutations resulting in AML. The characteristics of the patients enrolled in this study are shown in Table 2.
mtDNA sequence variants in AML patients
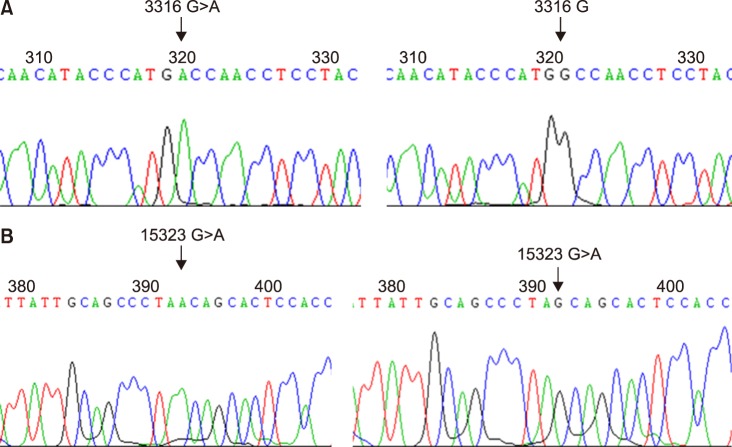
We established mtDNA polymorphism databases by analysis of HV1 and HV2 control regions and the tRNAleu1 and CYTB genes. Representative sequencing chromatograms revealing mtDNA mutations are shown in Fig. 1. In AML patients, a total number of 963 variants at 134 different positions were found, with a median of 12 variants per patient (Supplementary Table 1). In control subjects, 920 variants at 113 different positions were found, and the median of variants was 13. Most variations were single nucleotide substitutions, and only a small portion of insertions/deletions was detected (Supplementary Table 1). In patients with AML, 57 mtDNA variants were identified, which were absent in control subjects. We detected two novel variants, A16162T and G366A. The A16162T variant was found in three patients with AML (4.1%) and two control subjects (2.9%). G366A was observed only in one patient with AML (Supplementary Table 1). The variants of G16129A, A16182C, A16183C, T16189C, T489C, and G3010A showed statistically significant differences between patients with AML and control subjects. However, all five variants in HV1 and the variation in tRNAleu1 appeared more frequently in control subjects than in patients with AML, and only the T489C variant in HV2 was dominant in patients with AML.
Distribution of mtDNA minisatellite instability (mtMSI)
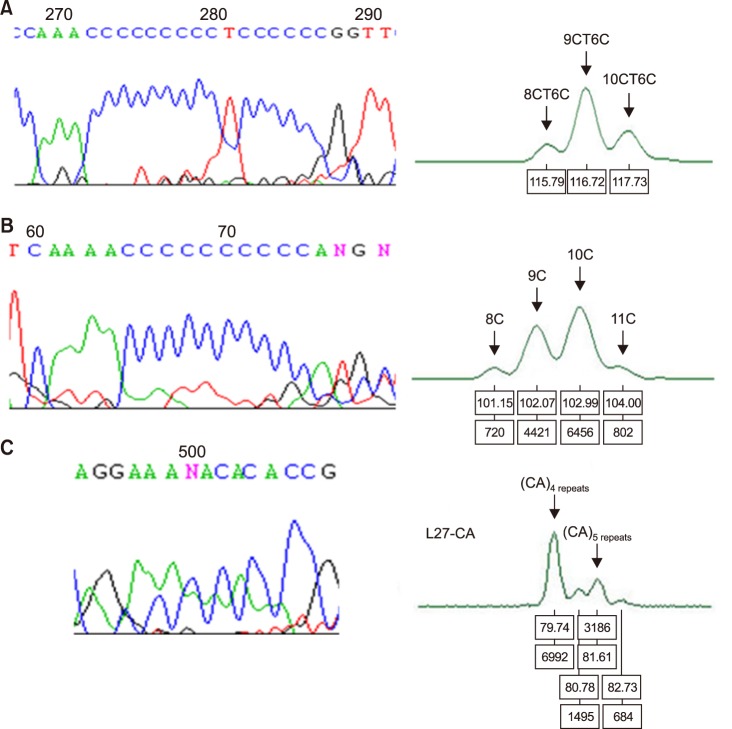
The overall frequency of mtMSI in patients with AML was 100% (N=74). Gene scan analyses of poly C-stretch region at nucleotide position (np) 303–315, 16184–16193, and 51–515(CA)5 repeats are shown in Fig. 2. All patients with AML had mtMSI values of 303 poly-C, and 70.3% (N=52) of the patients had mtMSIs in 16189 poly-C (Table 3). In our previous report of healthy Korean donors, the pattern number was 6 in 303 poly-C and 8 in 16189 poly-C [6]. However, in this study, the number of the patterns increased into 11 and 11, respectively.
mtDNA copy number
The mean mtDNA copy number of 70 control subjects was 45.1, and the range was from 3.6 to 142.3. Among the 74 patients with AML involved in this study, the copy number analysis was not available in six patients. The mean mtDNA copy number of 68 patients with AML was 503.1, and the range was from 4.0 to 7901.7. The mean mtDNA copy number of patients with AML increased approximately nine-fold compared to normal control subjects (P<0.0001).
mtDNA haplogroup in patients with AML
There were no statistically significant differences in the distribution of mtDNA haplogroup frequencies between patients with AML and control subjects, except for haplogroup D4 (Supplementary Table 2). Haplogroup D4 was found to have a higher frequency in patients with AML, 31.0% versus 15.7% (P=0.046), as compared to that in control subjects. This difference translated into an OR of 2.408 for AML (95% CI, 1.064–5.452).
Impact of mtDNA sequence variant, mtMSI, mtDNA copy number, and haplogroup on outcomes in patients with AML
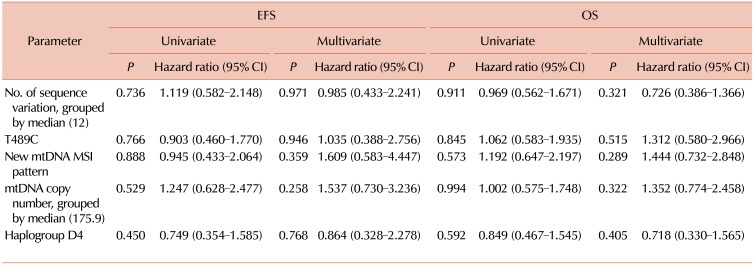
Fifty-seven of 74 patients were treated at our hospital, and 40 (70.2%) patients achieved CR with an induction of chemotherapy or underwent stem cell transplantation. Twenty-one (52.5%) patients relapsed or died after achieving remission. According to the aforementioned results, patients with a higher number (above the median) of sequence variants, T489C sequence alteration, new mtDNA MSI patterns, higher copy number (above the median), or haplogroup D4 were included to estimate the HR of EFS and OS. However, the survival data analyzed by Cox regression (univariate or multivariate) showed no statistical significance (Table 4).
DISCUSSION
Several studies on mtDNA genome variations in acute leukemia have been reported. He et al. [26] compared the entire mitochondrial genome from both normal tissue and leukemic samples from 24 leukemia patients. Grist et al. [19] sequenced the D loop (nt 16111–190) of 48 patients with leukemia. Yao et al. [27] analyzed the mtDNA control region sequence variation in single cells from 18 leukemia patients. Sharawat et al. [21] evaluated the entire D loop region in 44 pediatric patients with AML by direct sequencing. Silkjaer et al. [22] investigated the entire mtDNA in 56 AML patients. Finally, Han et al. [28] analyzed mutations and mtMSI in the mtDNA D-loop region in BM cells of 19 patients with acute leukemia.
In this study, we collected 74 patients with AML and 70 control subjects, and studied not only the mtDNA sequence variants, but also the mtMSI, copy number, and haplogroup. The median number of the mtDNA sequence variant was not different in the patients with AML and control subjects. The changed sequence also showed no significant difference between the two groups. He et al. [26] demonstrated that somatic mtDNA mutations were present in approximately 40% of patients, and they found the A15296G mutation as a leukemia-specific marker. In our study, no participants had a mutation on nucleotide (nt) 15296. Silkjaer et al. [22] reported that 21% of the AML patients harbored the T16311C variant, and this variant tended to be associated with chromosomal abnormalities of favorable prognosis. In this study, we found the T16311C variant in six control subjects (8.5%) and three patients with AML (4%). Among the three patients with AML, two patients had normal karyotypes and one had t(15;17). However, the sample size was too small to analyze for the statistical significance of the relationship and chromosome abnormality. We also found the T489C variant, which appeared in a higher proportion (73%) of patients with AML than in control subjects (50%), but it showed no impact on outcomes. All patients of the haplogroup D4 had the T489C variant.
In the present study, most patients with AML had mtMSI in 303 poly-C and 16189 poly-C (Table 3), and the pattern number of the mtMSI was greater in patients with AML than in control cohorts. These greater heteroplasmy lengths in patients with AML might be associated with the clinical implication. Mitochondrial genomic instability has been strongly correlated with a high incidence of circular mtDNA molecules (of multiple lengths) in human leukemic cells, and a positive correlation has been observed between the frequency of these molecules and the disease severity [29].
Meanwhile, Grist et al. [19] found mtDNA mutations in 36% of AML patients. Yao et al. [27] found that some patients at relapse presented a complex shift in major haplotypes. In our study, the haplogroup D4 was found at a higher frequency in patients with AML compared to that in control subjects, and this difference resulted in an OR of 2.408 for AML (P=0.046). Moreover, in the report by Sharawat et al. [21], some variations in HV-1 were associated with inferior EFS. Silkjaer et al. [22] suggested that alterations in cytochrome c oxidase subunit I and II have an adverse impact on prognosis. In addition, they reported that the T16311C variant tended to be associated with chromosomal abnormalities.
There are also limitations in this study. Since we compared the mtDNA from patients with AML with that from control subjects, there could be the possibility that the sequence changes between the patients with AML and controls may simply reflect extensive sequence variation within the population [30]. As shown in this study, there were no significant differences in mtDNA sequence variation between patients with AML and control subjects. However, even though the variants could not be compared with respect to the potential role in malignancy formation, mtDNA mutations might still contribute to genomic instability.
In conclusion, AML cells revealed more heterogeneous patterns in mtMSI markers and had increased mtDNA copy numbers. These findings implicate mitochondrial genome instability in primary AML cells. In addition, the mtDNA haplogroup D4 might be associated with AML risk among Koreans.

 XML Download
XML Download